Parte 1: Executar um pipeline de demonstração¶
Tradução assistida por IA - saiba mais e sugira melhorias
Nesta primeira parte do curso de treinamento Hello nf-core, mostramos como encontrar e experimentar um pipeline do nf-core, configurar e personalizar sua execução para suas necessidades, e entender como a validação de entrada protege contra erros comuns.
Vamos usar um pipeline chamado nf-core/demo que é mantido pelo projeto nf-core como parte de seu inventário de pipelines para fins de demonstração e treinamento.
Certifique-se de que seu diretório de trabalho esteja definido como hello-nf-core/ conforme instruído na página Primeiros passos.
1. Encontrar e recuperar o pipeline nf-core/demo¶
Vamos começar localizando o pipeline nf-core/demo no site do projeto em nf-co.re, que centraliza todas as informações, como: documentação geral e artigos de ajuda, documentação para cada um dos pipelines, posts de blog, anúncios de eventos e assim por diante.
1.1. Encontrar o pipeline no site¶
No seu navegador web, vá para https://nf-co.re/pipelines/ e digite demo na barra de pesquisa.
Clique no nome do pipeline, demo, para acessar a página de documentação do pipeline.
Cada pipeline lançado tem uma página dedicada que inclui as seguintes seções de documentação:
- Introduction: Uma introdução e visão geral do pipeline
- Usage: Descrições de como executar o pipeline
- Parameters: Parâmetros do pipeline agrupados com descrições
- Output: Descrições e exemplos dos arquivos de saída esperados
- Results: Exemplos de arquivos de saída gerados a partir do conjunto de dados de teste completo
- Releases & Statistics: Histórico de versões do pipeline e estatísticas
Sempre que você estiver considerando adotar um novo pipeline, você deve ler a documentação do pipeline cuidadosamente primeiro para entender o que ele faz e como deve ser configurado antes de tentar executá-lo.
Dê uma olhada agora e veja se você consegue descobrir:
- Quais ferramentas o pipeline executará (Verifique a aba:
Introduction) - Quais entradas e parâmetros o pipeline aceita ou requer (Verifique a aba:
Parameters) - Quais são as saídas produzidas pelo pipeline (Verifique a aba:
Output)
1.1.1. Visão geral do pipeline¶
A aba Introduction fornece uma visão geral do pipeline, incluindo uma representação visual (chamada de mapa de metrô) e uma lista de ferramentas que são executadas como parte do pipeline.

- Read QC (FASTQC)
- Adapter and quality trimming (SEQTK_TRIM)
- Present QC for raw reads (MULTIQC)
- Generate a lighthearted text message from a cow (COWPY)
1.1.2. Exemplo de linha de comando¶
A documentação também fornece um arquivo de entrada de exemplo (discutido mais adiante) e um exemplo de linha de comando.
nextflow run nf-core/demo \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR>
Você notará que o comando de exemplo NÃO especifica um arquivo de fluxo de trabalho, apenas a referência ao repositório do pipeline, nf-core/demo.
Quando invocado dessa forma, o Nextflow assumirá que o código está organizado de uma certa maneira. Vamos recuperar o código para que possamos examinar essa estrutura.
1.2. Recuperar o código do pipeline¶
Depois de determinarmos que o pipeline parece ser adequado para nossos propósitos, vamos experimentá-lo. Felizmente, o Nextflow facilita a recuperação de pipelines de repositórios formatados corretamente sem precisar baixar nada manualmente.
1.2.1. Usar nextflow pull¶
Vamos retornar ao terminal e executar o seguinte:
Saída do comando
O Nextflow faz um pull do código do pipeline, o que significa que ele baixa o repositório completo para sua unidade local.
Para ser claro, você pode fazer isso com qualquer pipeline Nextflow que esteja configurado adequadamente no GitHub, não apenas pipelines do nf-core. No entanto, o nf-core é a maior coleção de código aberto de pipelines Nextflow.
1.2.2. Usar nextflow list¶
Você pode fazer o Nextflow fornecer uma lista de quais pipelines você recuperou dessa maneira:
Você pode tentar fazer o pull de alguns outros pipelines para ver como eles aparecem na lista quando você tem mais de um.
1.2.3. Encontrar onde o pipeline foi baixado¶
Você notará que os arquivos não estão no seu diretório de trabalho atual.
Por padrão, o Nextflow salva os pipelines baixados em $NXF_HOME/assets.
Para encontrar onde um pipeline específico está localizado, pergunte diretamente ao Nextflow:
Saída do comando
project name: nf-core/demo
repository : https://github.com/nf-core/demo
local path : /workspaces/.nextflow/assets/.repos/nf-core/demo
main script : main.nf
description : An nf-core demo pipeline
revisions :
TEMPLATE
bumper
dev
fix-nxfversion
manually-merge-3_0_2
> master (default)
nf-core-template-merge-2.13.2.dev0
nf-core-template-merge-2.14.0
nf-core-template-merge-2.14.1
nf-core-template-merge-3.0.0
nf-core-template-merge-3.0.1
nf-core-template-merge-3.0.2
nf-core-template-merge-3.1.0
nf-core-template-merge-3.1.2
nf-core-template-merge-3.2.0
nf-core-template-merge-3.2.1
nf-core-template-merge-3.3.1
nf-core-template-merge-3.3.2
nf-core-template-merge-4.0.0
1.0.0 [t]
1.0.1 [t]
1.0.2 [t]
1.1.0 [t]
> 1.2.0 [t]
Info
O caminho completo pode ser diferente no seu sistema se você não estiver usando nosso ambiente de treinamento.
O Nextflow mantém o código-fonte baixado intencionalmente 'fora do caminho' com base no princípio de que esses pipelines devem ser usados mais como bibliotecas do que código com o qual você interagiria diretamente.
Internamente, o Nextflow armazena cada pipeline baixado como um repositório git em $NXF_HOME/assets/.repos/, e faz o checkout do código de cada revisão em um subdiretório clones/<commit>/.
Como .repos é um diretório oculto, um simples tree -L 2 $NXF_HOME/assets/ parecerá vazio.
1.2.4. Criar um link simbólico para acessar o código-fonte facilmente¶
Não vamos examinar o código em detalhes, mas vamos dar uma rápida olhada apenas para ter uma ideia de como é a organização geral.
Para facilitar a navegação pelo código-fonte do pipeline, crie um link simbólico apontando para a cópia do pipeline que foi baixada:
mkdir -p pipelines/nf-core
ln -s "$(echo $NXF_HOME/assets/.repos/nf-core/demo/clones/*/)" pipelines/nf-core/demo
Isso cria um atalho para que você possa explorar o código com tree -L 2 pipelines/nf-core/demo ou abrir arquivos diretamente.
1.2.5. Visão geral da organização do código¶
Você pode usar tree ou usar o explorador de arquivos para encontrar e abrir o diretório nf-core/demo.
Conteúdo do diretório
pipelines/nf-core/demo
├── assets
├── CHANGELOG.md
├── CITATIONS.md
├── CODE_OF_CONDUCT.md
├── conf
├── docs
├── LICENSE
├── main.nf
├── modules
├── modules.json
├── nextflow.config
├── nextflow_schema.json
├── nf-test.config
├── README.md
├── ro-crate-metadata.json
├── subworkflows
├── tests
├── tower.yml
└── workflows
7 directories, 12 files
Como você pode ver, há muita coisa acontecendo lá, mas a maior parte não precisa ser motivo de preocupação.
Brevemente, vamos observar que no nível superior, você pode encontrar um arquivo README com informações resumidas, bem como arquivos acessórios que resumem informações do projeto, como licenciamento, diretrizes de contribuição, citação e código de conduta.
A documentação detalhada do pipeline está localizada no diretório docs.
Todo esse conteúdo é usado para gerar as páginas da web no site do nf-core programaticamente, então elas estão sempre atualizadas com o código.
Para o restante, podemos distinguir três grupos funcionais de arquivos de código:
- Componentes de código do pipeline (
main.nf,workflows,subworkflows,modules) - Configuração do pipeline
- Parâmetros do pipeline / entradas e validação
Não vamos abordar os componentes de código do pipeline nesta parte do curso, mas vamos tocar em elementos de configuração e validação que provavelmente serão relevantes para você como usuário final de pipelines do nf-core.
Dica
Você também pode navegar pelo código-fonte de qualquer pipeline do nf-core no GitHub, por exemplo, github.com/nf-core/demo. Todo pipeline do nf-core segue o mesmo layout de diretório, então uma vez que você conheça a estrutura, pode encontrar arquivos de configuração, módulos e fluxos de trabalho para qualquer pipeline da mesma forma.
Mas por enquanto, vamos executar o pipeline!
Conclusão¶
Agora você sabe como encontrar um pipeline através do site do nf-core e recuperar uma cópia local do código-fonte.
O que vem a seguir?¶
Aprenda como experimentar um pipeline do nf-core com o mínimo de esforço.
2. Experimentar o pipeline com seu perfil de teste¶
Convenientemente, todo pipeline do nf-core vem com um perfil de teste. Este é um conjunto mínimo de configurações para o pipeline executar usando um pequeno conjunto de dados de teste hospedado no repositório nf-core/test-datasets. É uma ótima maneira de experimentar rapidamente um pipeline em pequena escala.
Dica
O sistema de perfil de configuração do Nextflow permite que você alterne facilmente entre diferentes motores de contêiner ou ambientes de execução. Para mais detalhes, consulte Hello Nextflow Parte 6: Configuração.
2.1. Examinar o perfil de teste¶
É uma boa prática verificar o que o perfil de teste de um pipeline especifica antes de executá-lo.
O perfil test para nf-core/demo está no arquivo de configuração conf/test.config.
Você pode encontrá-lo localmente dentro do código-fonte do pipeline que o nextflow pull baixou, através do link simbólico pipelines criado na seção 1.2.4:
Aqui está o conteúdo desse arquivo:
Você notará imediatamente que o bloco de comentário no topo inclui um exemplo de uso mostrando como executar o pipeline com este perfil de teste.
| conf/test.config | |
|---|---|
As únicas coisas que precisamos fornecer são o que é mostrado entre colchetes angulares no comando de exemplo: <docker/singularity> e <OUTDIR>.
Como lembrete, <docker/singularity> refere-se à escolha do sistema de contêiner. Todos os pipelines do nf-core são projetados para serem usáveis com contêineres (Docker, Singularity, etc.) para garantir reprodutibilidade e eliminar problemas de instalação de software.
Então precisaremos especificar se queremos usar Docker ou Singularity para testar o pipeline.
A parte --outdir <OUTDIR> refere-se ao diretório onde o Nextflow escreverá as saídas do pipeline.
Precisamos fornecer um nome para ele, que podemos simplesmente inventar.
Se ainda não existir, o Nextflow o criará para nós em tempo de execução.
Seguindo para a seção após o bloco de comentário, o perfil de teste nos mostra o que foi pré-configurado para teste: mais notavelmente, o parâmetro input já está configurado para apontar para um conjunto de dados de teste, então não precisamos fornecer nossos próprios dados.
Se você seguir o link para a entrada pré-configurada, verá que é um arquivo CSV contendo identificadores de amostra e caminhos de arquivo para várias amostras experimentais.
sample,fastq_1,fastq_2
SAMPLE1_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R2.fastq.gz
SAMPLE2_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R2.fastq.gz
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,
Isso é chamado de planilha de amostras e é a forma mais comum de entrada para pipelines do nf-core. Não se preocupe se você não estiver familiarizado com os formatos e tipos de dados, isso não é importante para o que se segue.
Agora temos tudo o que precisamos para experimentar o pipeline.
2.2. Executar o pipeline¶
Conforme mencionado acima, podemos usar o comando de teste de exemplo quase como está; só precisamos especificar qual sistema de empacotamento de software usar e como nomear o diretório de saída.
Aqui usaremos Docker para o sistema de contêiner e demo-results, respectivamente.
Com isso, podemos executar o comando de teste:
Saída do comando
N E X T F L O W ~ version 26.04.4
Downloading plugin nf-schema@2.7.2
Launching `https://github.com/nf-core/demo` [cranky_curry] revision: 32893afef8 [master]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.2.0
------------------------------------------------------
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : demo-results
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Generic options
trace_report_suffix : 2026-07-03_21-31-35
Core Nextflow options
revision : master
runName : cranky_curry
containerEngine : docker
launchDir : /workspaces/training/hello-nf-core
workDir : /workspaces/training/hello-nf-core/work
projectDir : /workspaces/.nextflow/assets/.repos/nf-core/demo/clones/32893afef8076a03a2767a020b3f0cab2e0b40b2
userName : root
profile : docker,test
configFiles : /workspaces/.nextflow/assets/.repos/nf-core/demo/clones/32893afef8076a03a2767a020b3f0cab2e0b40b2/nextflow.config
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
executor > local (8)
[ca/5b0f3e] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) [100%] 3 of 3 ✔
[b7/cb6812] NFCORE_DEMO:DEMO:SEQTK_TRIM (SAMPLE3_SE) [100%] 3 of 3 ✔
[ff/6ebd98] NFCORE_DEMO:DEMO:COWPY [100%] 1 of 1 ✔
[09/bbd1b4] NFCORE_DEMO:DEMO:MULTIQC (demo) [100%] 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
Se sua saída corresponder a isso, parabéns! Você acabou de executar seu primeiro pipeline do nf-core.
Você notará que há muito mais saída no console do que quando você executa um pipeline Nextflow básico. Há um cabeçalho que inclui um resumo da versão do pipeline, entradas e saídas, e alguns elementos de configuração.
Info
Sua saída mostrará carimbos de data/hora, nomes de execução e caminhos de arquivo diferentes, mas a estrutura geral e a execução do processo devem ser semelhantes.
Observe a linha próxima ao topo da saída:
Isso informa qual revisão do pipeline foi usada.
Como não especificamos uma versão, o Nextflow usou o commit mais recente no master.
Para execuções reproduzíveis, você deve fixar uma versão específica usando a flag -r:
Isso garante que o mesmo código do pipeline seja usado sempre, independentemente de novos commits ou lançamentos.
Para este treinamento, omitimos -r por simplicidade, mas em produção você deve sempre especificá-lo.
Seguindo para a saída de execução, vamos dar uma olhada nas linhas que nos dizem quais processos foram executados:
executor > local (8)
[ca/5b0f3e] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) [100%] 3 of 3 ✔
[b7/cb6812] NFCORE_DEMO:DEMO:SEQTK_TRIM (SAMPLE3_SE) [100%] 3 of 3 ✔
[ff/6ebd98] NFCORE_DEMO:DEMO:COWPY [100%] 1 of 1 ✔
[09/bbd1b4] NFCORE_DEMO:DEMO:MULTIQC (demo) [100%] 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
Isso nos diz que quatro processos foram executados, correspondendo às quatro ferramentas mostradas na página de documentação do pipeline no site do nf-core: FASTQC, SEQTK_TRIM, MULTIQC e COWPY.
Os nomes completos dos processos como mostrado aqui, como NFCORE_DEMO:DEMO:MULTIQC, são mais longos do que o que você pode ter visto no material introdutório do Hello Nextflow.
Estes incluem os nomes de seus fluxos de trabalho pai e refletem a modularidade do código do pipeline.
Vamos entrar em mais detalhes sobre isso na Parte 2 deste curso.
2.3. Examinar as saídas do pipeline¶
Finalmente, vamos dar uma olhada no diretório demo-results produzido pelo pipeline.
Conteúdo do diretório
demo-results
├── cowpy
│ └── cowpy.txt
├── fastqc
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── fq
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── multiqc
│ ├── multiqc_data
│ └── multiqc_report.html
└── pipeline_info
├── execution_report_2026-07-03_21-31-35.html
├── execution_timeline_2026-07-03_21-31-35.html
├── execution_trace_2026-07-03_21-31-35.txt
├── nf_core_demo_software_mqc_versions.yml
├── params_2026-07-03_21-31-43.json
└── pipeline_dag_2026-07-03_21-31-35.html
12 directories, 8 files
Isso pode parecer muito.
Para saber mais sobre as saídas do pipeline nf-core/demo, consulte sua página de documentação.
Nesta etapa, o que é importante observar é que os resultados são organizados por módulo, e há adicionalmente um diretório chamado pipeline_info contendo vários relatórios com carimbos de data/hora sobre a execução do pipeline.
Por exemplo, o arquivo execution_timeline_* mostra quais processos foram executados, em que ordem e quanto tempo levaram para executar:
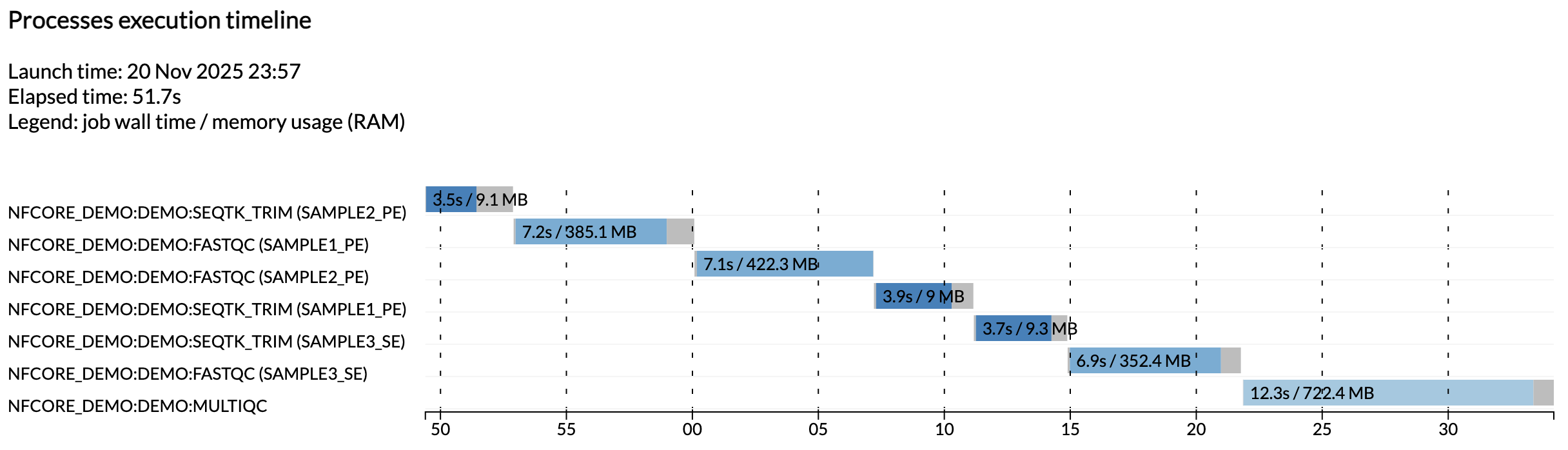
Info
Aqui as tarefas não foram executadas em paralelo porque estamos executando em uma máquina minimalista no Github Codespaces. Para ver essas execuções em paralelo, tente aumentar a alocação de CPU do seu codespace e os limites de recursos na configuração de teste.
Esses relatórios são gerados automaticamente para todos os pipelines do nf-core.
Conclusão¶
Você sabe como executar um pipeline do nf-core usando seu perfil de teste integrado e onde encontrar suas saídas.
O que vem a seguir?¶
Aprenda como configurar o pipeline para personalizar sua execução.
3. Configurar a execução do pipeline¶
Conforme explicado em Hello Config, queremos poder alterar em quais dados nosso pipeline será executado e como ele será executado sem alterar o código do pipeline em si. Para isso, o Nextflow suporta múltiplas formas de controlar a configuração do pipeline, o que pode ser um pouco avassalador.
O projeto nf-core especifica convenções para organizar elementos de configuração, distinguindo dois tipos de configuração no nível superior: parâmetros do pipeline e configuração no sentido estrito.
- Parâmetros do pipeline (definidos através do sistema
params) tipicamente incluem coisas como arquivos de entrada, flags de comportamento de ferramentas e parâmetros de análise. - Configuração no sentido estrito refere-se à logística de como o pipeline é executado, ou seja, o executor, alocações de recursos computacionais e assim por diante.
Vamos começar abordando os parâmetros do pipeline e depois veremos a configuração no sentido estrito.
3.1. Parâmetros do pipeline¶
Para todos os pipelines do nf-core, você pode obter uma lista completa de parâmetros do pipeline diretamente da linha de comando usando a flag --help, que é ela própria um parâmetro do pipeline.
3.1.1. Obter a lista de parâmetros com --help¶
Execute o comando de ajuda para o pipeline de demonstração:
Saída do comando
N E X T F L O W ~ version 26.04.4
Launching `https://github.com/nf-core/demo` [adoring_meucci] revision: 32893afef8 [master]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.2.0
------------------------------------------------------
Typical pipeline command:
nextflow run nf-core/demo -profile <docker/singularity/.../institute> --input samplesheet.csv --outdir <OUTDIR>
Input/output options
--input [string] Path to a metadata file containing information about the samples in the experiment.
--outdir [string] The output directory where the results will be saved. You have to use absolute paths to storage on Cloud infrastructure.
--email [string] Email address for completion summary.
--multiqc_title [string] MultiQC report title. Printed as page header, used for filename if not otherwise specified.
Reference genome options
--genome [string] Name of iGenomes reference.
--fasta [string] Path to FASTA genome file.
Process skipping options
--skip_trim [boolean] Skip trimming fastq files with seqtk
Generic options
--multiqc_methods_description [string] Custom MultiQC yaml file containing HTML including a methods description.
--help [boolean, string] Display the help message.
--help_full [boolean] Display the full detailed help message.
--show_hidden [boolean] Display hidden parameters in the help message (only works when --help or --help_full are provided).
!! Hiding 19 param(s), use the `--showHidden` parameter to show them !!
------------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
Como você pode ver, a saída agrupa os parâmetros em categorias (opções de entrada/saída, opções de genoma de referência, etc.) com tipos e descrições para cada um.
Essa categorização é determinada por um arquivo de schema, que é abordado mais adiante.
Em pipelines Nextflow simples, --help só funciona se o desenvolvedor o implementou manualmente.
Dica
Use --help --show_hidden para ver parâmetros adicionais que estão ocultos por padrão, como --publish_dir_mode ou --monochrome_logs.
3.1.2. Definir valores de parâmetros¶
Conforme abordado em Hello Config, você pode definir valores de parâmetros na linha de comando com --nome_do_param ou coletar um conjunto de parâmetros em um arquivo YAML e passá-lo com -params-file.
Ambas as abordagens funcionam da mesma forma com pipelines do nf-core.
Por exemplo, para pular a etapa de trimming, queremos definir o parâmetro boolean skip_trim como true.
Um arquivo de parâmetros chamado my_params.yml está disponível no seu diretório de trabalho com esse valor já definido:
Passe-o com -params-file:
nextflow run nf-core/demo -profile docker,test --outdir demo-results-notrim -params-file my_params.yml
Saída do comando
N E X T F L O W ~ version 26.04.4
Launching `https://github.com/nf-core/demo` [focused_heisenberg] revision: 32893afef8 [master]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.2.0
------------------------------------------------------
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : demo-results-notrim
Process skipping options
skip_trim : true
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Generic options
trace_report_suffix : 2026-07-03_22-08-47
Core Nextflow options
revision : master
runName : focused_heisenberg
containerEngine : docker
launchDir : /workspaces/training/hello-nf-core
workDir : /workspaces/training/hello-nf-core/work
projectDir : /workspaces/.nextflow/assets/.repos/nf-core/demo/clones/32893afef8076a03a2767a020b3f0cab2e0b40b2
userName : root
profile : docker,test
configFiles : /workspaces/.nextflow/assets/.repos/nf-core/demo/clones/32893afef8076a03a2767a020b3f0cab2e0b40b2/nextflow.config
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
executor > local (5)
[7a/f3599e] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) [100%] 3 of 3 ✔
[b0/2f0bdc] NFCORE_DEMO:DEMO:COWPY [100%] 1 of 1 ✔
[c3/3c2278] NFCORE_DEMO:DEMO:MULTIQC (demo) [100%] 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
O processo SEQTK_TRIM não aparece mais na saída.
Aviso: Limitações importantes sobre entradas de parâmetros
Definindo parâmetros boolean na linha de comando
A partir da versão 26.04 do Nextflow, todos os valores fornecidos na linha de comando são tipados como strings.
Para um parâmetro boolean como skip_trim, passá-lo como uma flag simples (--skip_trim) ou como --skip_trim true é avaliado como a string "true", o que falha na validação do schema:
Para definir um parâmetro boolean com um valor genuíno true/false, use um -params-file como mostrado acima, ou defina-o em um arquivo de configuração.
Parâmetros do tipo string, integer e file-path não são afetados e ainda podem ser definidos diretamente na linha de comando.
Este curso usa esse padrão ao longo de todo o material para parâmetros boolean.
Usando arquivos de configuração personalizados
Embora seja tecnicamente possível definir parâmetros do pipeline em um arquivo de configuração personalizado passado com -c, isso pode não substituir os padrões já definidos no próprio nextflow.config do pipeline, dependendo das regras de precedência de configuração do Nextflow.
Usar --nome_do_param na linha de comando ou -params-file é mais confiável, pois estes sempre têm precedência.
Como regra geral: se aparece na saída do --help, defina-o via linha de comando ou um arquivo de parâmetros em vez de um arquivo de configuração.
3.1.3. Validação de parâmetros¶
Curiosidade: o comando --help funciona para todos os pipelines do nf-core porque o projeto nf-core exige que os desenvolvedores definam todos os parâmetros do pipeline formalmente em um arquivo de schema JSON (nextflow_schema.json).
Esse schema registra o tipo, descrição, valor padrão e agrupamento de cada parâmetro.
Além de alimentar a saída do --help, o arquivo de schema também permite a validação automatizada no momento do lançamento.
Isso significa que o Nextflow pode verificar se cada parâmetro que você passa existe e recebeu um valor apropriado (do tipo correto, dentro do intervalo de valores permitidos, etc.).
Abordamos isso com mais detalhes na Parte 5: Validação de Entrada, mas você já pode ver isso em ação fornecendo ao pipeline de demonstração alguma entrada de parâmetro inválida.
3.1.3.1. Parâmetros não reconhecidos¶
Tente passar um parâmetro que não existe:
A saída do console inclui um aviso:
O pipeline ainda é executado, mas o aviso alerta imediatamente que --foobar não é um parâmetro reconhecido.
Isso serve para chamar sua atenção para erros de digitação que não causam falha, como usar --outDir em vez de --outdir, o que pode ajudá-lo a evitar desperdício de tempo e recursos computacionais.
3.1.3.2. Valores de parâmetros inválidos¶
A validação também verifica os valores dos parâmetros.
O parâmetro --skip_trim é uma flag boolean, então passar um valor string faz o pipeline falhar imediatamente:
ERROR ~ Validation of pipeline parameters failed!
-- Check '.nextflow.log' file for details
The following invalid input values have been detected:
* --skip_trim (yes): Value is [string] but should be [boolean]
O pipeline para antes que qualquer processo seja executado, evitando uma execução com falha ou incorreta.
Conforme mencionado na seção 3.1.2, parâmetros boolean devem ser definidos com um valor genuíno true/false em um arquivo de parâmetros em vez de serem passados na linha de comando, pois os valores da linha de comando são tipados como strings.
3.1.4. Validação de entrada¶
A mesma lógica de validação também pode ser usada para verificar a validade dos arquivos de entrada. Por exemplo, se um pipeline espera uma planilha de amostras como sua principal entrada de dados (o que é o caso de muitos, se não da maioria dos pipelines do nf-core), o desenvolvedor pode fornecer um schema de entrada (distinto do schema de parâmetros) descrevendo como o arquivo de entrada deve ser estruturado.
Então, em tempo de execução, o Nextflow pode verificar se o arquivo de entrada fornecido é válido.
Também abordamos isso com mais detalhes na Parte 5: Validação de Entrada, mas você já pode ver isso em ação fornecendo ao pipeline de demonstração uma planilha de amostras inválida.
O pipeline nf-core/demo espera um arquivo CSV com as colunas sample, fastq_1 e fastq_2.
Isso é definido em um arquivo de schema (assets/schema_input.json) que especifica a estrutura esperada, tipos de coluna e restrições.
Arquivo de schema para entradas
{
"$schema": "https://json-schema.org/draft/2020-12/schema",
"$id": "https://raw.githubusercontent.com/nf-core/demo/master/assets/schema_input.json",
"title": "nf-core/demo pipeline - params.input schema",
"description": "Schema for the file provided with params.input",
"type": "array",
"items": {
"type": "object",
"properties": {
"sample": {
"type": "string",
"pattern": "^\\S+$",
"errorMessage": "Sample name must be provided and cannot contain spaces",
"meta": ["id"]
},
"fastq_1": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 1 must be provided, cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
},
"fastq_2": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 2 cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
}
},
"required": ["sample", "fastq_1"]
}
}
O schema especifica que sample e fastq_1 são obrigatórios, enquanto fastq_2 é opcional (suportando dados paired-end e single-end).
Os caminhos de arquivo são validados quanto à existência e padrão de extensão.
Para demonstrar isso, fornecemos uma planilha de amostras malformada chamada malformed_samplesheet.csv no seu diretório de trabalho:
Esta planilha de amostras está faltando a coluna obrigatória fastq_1 e tem um caminho de arquivo inexistente em fastq_2.
Execute o pipeline de demonstração usando malformed_samplesheet.csv como entrada:
nextflow run nf-core/demo -profile docker,test --outdir demo-results --input malformed_samplesheet.csv
ERROR ~ Validation of pipeline parameters failed!
-- Check '.nextflow.log' file for details
The following invalid input values have been detected:
* --input (malformed_samplesheet.csv): Validation of file failed:
-> Entry 1: Error for field 'fastq_2' (/not/a/real/file.fastq.gz): the file or directory
'/not/a/real/file.fastq.gz' does not exist (FastQ file for reads 2 cannot contain spaces
and must have extension '.fq.gz' or '.fastq.gz')
-> Entry 1: Missing required field(s): fastq_1
Como você pode ver, o pipeline falha imediatamente e relata todos os erros de validação de uma vez. O nf-schema não para no primeiro erro — ele coleta todos os problemas e os lista juntos, para que você possa corrigir tudo de uma vez em vez de descobrir os problemas um por um.
Cada erro identifica a entrada e o campo exatos que causaram o problema, para que você possa corrigir sua planilha de amostras e relançar o pipeline com a confiança de que ele não vai falhar em algum momento posterior quando o Nextflow for acessar o caminho do arquivo.
Para desenvolvedores, tudo isso é abordado com mais detalhes na Parte 5 deste curso.
3.2. Configuração¶
A configuração no sentido estrito controla como o pipeline é executado: alocação de recursos, argumentos específicos de ferramentas, onde as tarefas são executadas e qual sistema de empacotamento de software usar.
Os pipelines do nf-core incluem configuração padrão em nextflow.config e no diretório conf/.
Antes de substituir qualquer coisa, é útil saber onde ficam os padrões.
Você já viu na seção 2.1 que o código-fonte do pipeline está em $NXF_HOME/assets.
Usando o link simbólico pipelines da seção 1.2.4, liste os arquivos de configuração para ver o que está disponível:
base.config
containers_conda_lock_files_amd64.config
containers_conda_lock_files_arm64.config
containers_docker_amd64.config
containers_docker_arm64.config
containers_singularity_https_amd64.config
containers_singularity_https_arm64.config
containers_singularity_oras_amd64.config
containers_singularity_oras_arm64.config
igenomes.config
igenomes_ignored.config
modules.config
test.config
test_full.config
Os arquivos de configuração mais importantes são:
conf/base.config: Define labels de recursos (process_low,process_medium,process_high) que atribuem CPUs, memória e tempo aos processos. Quando você vê um processo usando mais recursos do que o esperado, é aqui que esses padrões vêm.conf/modules.config: Define argumentos de ferramentas por processo (ext.args) e configurações de publicação de saída (publishDir). Abra este arquivo para ver quais argumentos cada ferramenta recebe por padrão.conf/test.config: O perfil de teste que você usou na seção 2.1, que limita recursos viaresourceLimitse define uma planilha de amostras de teste. Ativado com-profile test. Há também umconf/test_full.configpara executar com um conjunto de dados de teste de tamanho completo, útil para benchmarking.
O nextflow.config central carrega todos os itens acima e define os valores padrão apropriados para tudo.
Se você deseja modificar qualquer uma das configurações especificadas nesses arquivos, não modifique nenhum deles diretamente.
Em vez disso, crie seu próprio arquivo de configuração e passe-o com -c.
Os valores que você especificar substituirão os valores padrão definidos nesses outros arquivos.
Vamos experimentar isso na prática.
3.2.1. Personalizar recursos de processos e argumentos de ferramentas¶
Os módulos do nf-core suportam dois tipos comuns de substituição de configuração: alocação de recursos (CPUs, memória, tempo) e argumentos de ferramentas via ext.args.
Muitas ferramentas de linha de comando têm argumentos que não são usados com frequência suficiente para serem expostos como parâmetros do pipeline.
A convenção ext.args permite que você passe esses argumentos para a ferramenta subjacente através de um arquivo de configuração.
O arquivo custom.config disponível no seu diretório de trabalho demonstra ambas as substituições:
| custom.config | |
|---|---|
O primeiro bloco substitui a alocação de recursos do FASTQC.
Por padrão, o FASTQC usa o label process_medium do base.config, que aloca 6 CPUs e 36 GB de memória; aqui limitamos a 2 CPUs e 4 GB.
O segundo bloco passa um argumento extra para o SEQTK_TRIM via ext.args.
A flag -b 5 instrui o seqtk trimfq a remover 5 bases do início de cada leitura, além do trimming por qualidade.
Execute o pipeline com esta configuração:
Saída do comando
A flag -c adiciona sua configuração sobre a configuração integrada do pipeline.
Para verificar se a substituição do ext.args teve efeito, encontre o hash do diretório de trabalho do SEQTK_TRIM na saída da execução (por exemplo, work/17/428668...) e verifique o arquivo .command.sh dentro dele:
Saída do comando
Você deve ver -b 5 no comando seqtk trimfq.
Uma coisa importante a saber sobre ext.args: se um módulo já tem um valor padrão definido, seu valor irá substituí-lo completamente em vez de ser adicionado a ele.
Por exemplo, o FASTQC tem ext.args = '--quiet' definido por padrão em conf/modules.config:
| conf/modules.config | |
|---|---|
Se você definir ext.args = '--kmers 8' para o FASTQC, a flag --quiet não será mais aplicada.
Para manter ambos, defina ext.args = '--quiet --kmers 8'.
Você deve sempre verificar a configuração padrão de um módulo antes de substituir ext.args.
Conclusão¶
Você sabe como obter ajuda de um pipeline do nf-core, definir parâmetros e entender como eles são validados, e personalizar a configuração através de arquivos de configuração.
O que vem a seguir?¶
Se você só quer executar pipelines do nf-core, você terminou!
Se você quer aprender a desenvolver seus próprios pipelines seguindo os padrões do nf-core, faça uma pausa e passe para a Parte 2 quando estiver pronto. Você aprenderá a criar seu próprio pipeline compatível com o nf-core usando as ferramentas baseadas no template do nf-core.