Teil 1: Eine Demo-Pipeline ausführen¶
KI-gestützte Übersetzung - mehr erfahren & Verbesserungen vorschlagen
Im ersten Teil des Hello nf-core Trainingskurses zeigen wir dir, wie du eine nf-core Pipeline findest und ausprobierst, ihre Ausführung für deine Bedürfnisse konfigurierst und anpasst, und wie die Eingabevalidierung vor häufigen Fehlern schützt.
Wir werden eine Pipeline namens nf-core/demo verwenden, die vom nf-core-Projekt als Teil seines Pipeline-Inventars für Demonstrations- und Trainingszwecke gepflegt wird.
Stelle sicher, dass dein Arbeitsverzeichnis auf hello-nf-core/ gesetzt ist, wie auf der Seite Erste Schritte beschrieben.
1. Die nf-core/demo Pipeline finden und abrufen¶
Beginnen wir damit, die nf-core/demo Pipeline auf der Projektwebsite unter nf-co.re zu finden, die alle Informationen zentralisiert, wie: allgemeine Dokumentation und Hilfe-Artikel, Dokumentation für jede der Pipelines, Blogbeiträge, Event-Ankündigungen und so weiter.
1.1. Die Pipeline auf der Website finden¶
Gehe in deinem Webbrowser zu https://nf-co.re/pipelines/ und tippe demo in die Suchleiste.
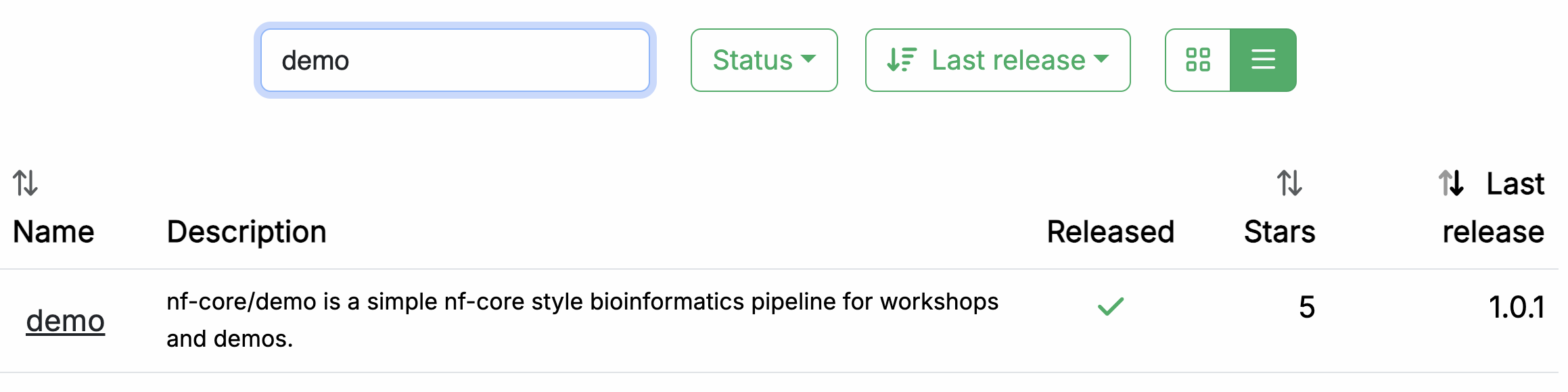
Klicke auf den Pipeline-Namen, demo, um auf die Pipeline-Dokumentationsseite zuzugreifen.
Jede veröffentlichte Pipeline hat eine eigene Seite, die die folgenden Dokumentationsabschnitte enthält:
- Introduction: Eine Einführung und Übersicht über die Pipeline
- Usage: Beschreibungen, wie die Pipeline ausgeführt wird
- Parameters: Gruppierte Pipeline-Parameter mit Beschreibungen
- Output: Beschreibungen und Beispiele der erwarteten Ausgabedateien
- Results: Beispiel-Ausgabedateien, die aus dem vollständigen Testdatensatz generiert wurden
- Releases & Statistics: Pipeline-Versionsverlauf und Statistiken
Wann immer du erwägst, eine neue Pipeline zu übernehmen, solltest du die Pipeline-Dokumentation zuerst sorgfältig lesen, um zu verstehen, was sie tut und wie sie konfiguriert werden sollte, bevor du versuchst, sie auszuführen.
Schau dir das jetzt an und sieh, ob du herausfinden kannst:
- Welche Tools die Pipeline ausführen wird (Prüfe den Tab:
Introduction) - Welche Eingaben und Parameter die Pipeline akzeptiert oder benötigt (Prüfe den Tab:
Parameters) - Was sind die Ausgaben, die von der Pipeline produziert werden (Prüfe den Tab:
Output)
1.1.1. Pipeline-Übersicht¶
Der Tab Introduction bietet eine Übersicht über die Pipeline, einschließlich einer visuellen Darstellung (genannt U-Bahn-Karte) und einer Liste von Tools, die als Teil der Pipeline ausgeführt werden.

- Read QC (FASTQC)
- Adapter and quality trimming (SEQTK_TRIM)
- Present QC for raw reads (MULTIQC)
1.1.2. Beispiel-Befehlszeile¶
Die Dokumentation bietet auch eine Beispiel-Eingabedatei (weiter unten ausführlicher besprochen) und eine Beispiel-Befehlszeile.
nextflow run nf-core/demo \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR>
Du wirst bemerken, dass der Beispielbefehl KEINE Workflow-Datei angibt, sondern nur die Referenz zum Pipeline-Repository, nf-core/demo.
Wenn es auf diese Weise aufgerufen wird, wird Nextflow annehmen, dass der Code auf eine bestimmte Weise organisiert ist. Lass uns den Code abrufen, damit wir diese Struktur untersuchen können.
1.2. Den Pipeline-Code abrufen¶
Nachdem wir festgestellt haben, dass die Pipeline für unsere Zwecke geeignet zu sein scheint, lass sie uns ausprobieren. Glücklicherweise macht es Nextflow einfach, Pipelines aus korrekt formatierten Repositories abzurufen, ohne irgendetwas manuell herunterladen zu müssen.
1.2.1. nextflow pull verwenden¶
Kehren wir zum Terminal zurück und führen Folgendes aus:
Befehlsausgabe
Nextflow führt einen pull des Pipeline-Codes durch, was bedeutet, dass es das vollständige Repository auf deine lokale Festplatte herunterlädt.
Um es klarzustellen: Du kannst dies mit jeder Nextflow-Pipeline tun, die entsprechend auf GitHub eingerichtet ist, nicht nur mit nf-core Pipelines. Allerdings ist nf-core die größte Open-Source-Sammlung von Nextflow-Pipelines.
1.2.2. nextflow list verwenden¶
Du kannst Nextflow veranlassen, dir eine Liste der Pipelines zu geben, die du auf diese Weise abgerufen hast:
Du kannst auch weitere Pipelines pullen, um zu sehen, wie sie aufgelistet werden, wenn du mehr als eine hast.
1.2.3. Deine Pipelines in $NXF_HOME/assets/ finden¶
Du wirst bemerken, dass die Dateien nicht in deinem aktuellen Arbeitsverzeichnis sind.
Standardmäßig speichert Nextflow sie unter $NXF_HOME/assets.
Hinweis
Der vollständige Pfad kann auf deinem System abweichen, wenn du nicht unsere Trainingsumgebung verwendest.
Nextflow hält den heruntergeladenen Quellcode absichtlich 'aus dem Weg' nach dem Prinzip, dass diese Pipelines eher wie Bibliotheken verwendet werden sollten als Code, mit dem du direkt interagieren würdest.
1.2.4. Einen Symlink erstellen, um leicht auf den Quellcode zuzugreifen¶
Wir werden den Code nicht im Detail durchgehen, aber lass uns kurz einen Blick darauf werfen, um ein Gefühl für die Gesamtorganisation zu bekommen.
Um das Durchsuchen des Pipeline-Quellcodes zu erleichtern, erstelle einen symbolischen Link zum Assets-Verzeichnis:
Dies erstellt eine Verknüpfung, mit der du den Code mit tree -L 2 pipelines erkunden oder Dateien direkt öffnen kannst.
1.2.5. Überblick über die Code-Organisation¶
Du kannst entweder tree verwenden oder den Datei-Explorer benutzen, um das nf-core/demo-Verzeichnis zu finden und zu öffnen.
Verzeichnisinhalt
pipelines/nf-core/demo
├── assets
├── CHANGELOG.md
├── CITATIONS.md
├── CODE_OF_CONDUCT.md
├── conf
├── docs
├── LICENSE
├── main.nf
├── modules
├── modules.json
├── nextflow.config
├── nextflow_schema.json
├── nf-test.config
├── README.md
├── ro-crate-metadata.json
├── subworkflows
├── tests
├── tower.yml
└── workflows
Wie du siehst, ist da eine Menge los, über das du dir aber größtenteils keine Gedanken machen musst.
Kurz gesagt: Auf der obersten Ebene findest du eine README-Datei mit zusammenfassenden Informationen sowie Hilfsdateien, die Projektinformationen wie Lizenzierung, Beitragsrichtlinien, Zitate und Verhaltenskodex zusammenfassen.
Detaillierte Pipeline-Dokumentation befindet sich im docs-Verzeichnis.
All dieser Inhalt wird verwendet, um die Webseiten auf der nf-core-Website programmatisch zu generieren, sodass sie immer mit dem Code auf dem neuesten Stand sind.
Für den Rest können wir drei funktionale Gruppen von Code-Dateien unterscheiden:
- Pipeline-Code-Komponenten (
main.nf,workflows,subworkflows,modules) - Pipeline-Konfiguration
- Pipeline-Parameter / Eingaben und Validierung
Wir werden die Pipeline-Code-Komponenten in diesem Teil des Kurses nicht durchgehen, aber wir werden Elemente der Konfiguration und Validierung ansprechen, die für dich als Endnutzer*in von nf-core Pipelines relevant sein dürften.
Tipp
Du kannst den Quellcode jeder nf-core Pipeline auch auf GitHub durchsuchen, z.B. github.com/nf-core/demo. Jede nf-core Pipeline folgt demselben Verzeichnislayout. Wenn du die Struktur einmal kennst, findest du Konfigurationsdateien, Module und Workflows für jede Pipeline auf die gleiche Weise.
Aber jetzt geht es darum, die Pipeline auszuführen!
Fazit¶
Du weißt jetzt, wie du eine Pipeline über die nf-core-Website finden und eine lokale Kopie des Quellcodes abrufen kannst.
Wie geht es weiter?¶
Lerne, wie du eine nf-core Pipeline mit minimalem Aufwand ausprobieren kannst.
2. Die Pipeline mit ihrem Testprofil ausprobieren¶
Praktischerweise kommt jede nf-core Pipeline mit einem Testprofil. Dies ist ein minimaler Satz von Konfigurationseinstellungen für die Pipeline, um mit einem kleinen Testdatensatz ausgeführt zu werden, der im nf-core/test-datasets Repository gehostet wird. Es ist eine großartige Möglichkeit, eine Pipeline schnell in kleinem Maßstab auszuprobieren.
Hinweis
Das Konfigurationsprofil-System von Nextflow ermöglicht es dir, einfach zwischen verschiedenen Container-Engines oder Ausführungsumgebungen zu wechseln. Für weitere Details siehe Hello Nextflow Teil 6: Konfiguration.
2.1. Das Testprofil untersuchen¶
Es ist gute Praxis, zu prüfen, was das Testprofil einer Pipeline spezifiziert, bevor man sie ausführt.
Das test-Profil für nf-core/demo befindet sich in der Konfigurationsdatei conf/test.config.
Du findest es lokal im Pipeline-Quellcode, den nextflow pull heruntergeladen hat:
Hier ist der Inhalt dieser Datei:
Du wirst sofort bemerken, dass der Kommentarblock oben ein Verwendungsbeispiel enthält, das zeigt, wie die Pipeline mit diesem Testprofil ausgeführt wird.
| conf/test.config | |
|---|---|
Die einzigen Dinge, die wir angeben müssen, sind das, was zwischen spitzen Klammern im Beispielbefehl gezeigt wird: <docker/singularity> und <OUTDIR>.
Zur Erinnerung: <docker/singularity> bezieht sich auf die Wahl des Container-Systems. Alle nf-core Pipelines sind darauf ausgelegt, mit Containern (Docker, Singularity, etc.) verwendbar zu sein, um Reproduzierbarkeit zu gewährleisten und Software-Installationsprobleme zu eliminieren.
Also müssen wir angeben, ob wir Docker oder Singularity verwenden möchten, um die Pipeline zu testen.
Der Teil --outdir <OUTDIR> bezieht sich auf das Verzeichnis, in das Nextflow die Ausgaben der Pipeline schreiben wird.
Wir müssen einen Namen dafür angeben, den wir einfach erfinden können.
Wenn es noch nicht existiert, wird Nextflow es zur Laufzeit für uns erstellen.
Weiter zum Abschnitt nach dem Kommentarblock zeigt uns das Testprofil, was für das Testen vorkonfiguriert wurde: am wichtigsten ist, dass der Parameter input bereits so eingestellt ist, dass er auf einen Testdatensatz zeigt, sodass wir keine eigenen Daten bereitstellen müssen.
Wenn du dem Link zur vorkonfigurierten Eingabe folgst, wirst du sehen, dass es sich um eine CSV-Datei handelt, die Probenidentifikatoren und Dateipfade für mehrere experimentelle Proben enthält.
sample,fastq_1,fastq_2
SAMPLE1_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R2.fastq.gz
SAMPLE2_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R2.fastq.gz
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,
Dies wird als Samplesheet bezeichnet und ist die häufigste Form der Eingabe für nf-core Pipelines.
Hinweis
Mach dir keine Sorgen, wenn du mit den Datenformaten und -typen nicht vertraut bist, es ist nicht wichtig für das Folgende.
Dies bestätigt also, dass wir alles haben, was wir brauchen, um die Pipeline auszuprobieren.
2.2. Die Pipeline ausführen¶
Entscheiden wir uns, Docker für das Container-System zu verwenden und demo-results als Ausgabeverzeichnis, und wir sind bereit, den Testbefehl auszuführen:
Befehlsausgabe
N E X T F L O W ~ version 25.10.4
Launching `https://github.com/nf-core/demo` [magical_pauling] DSL2 - revision: 45904cb9d1 [master]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.1.0
------------------------------------------------------
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : demo-results
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Generic options
trace_report_suffix : 2025-11-21_04-57-41
Core Nextflow options
revision : master
runName : magical_pauling
containerEngine : docker
launchDir : /workspaces/training/hello-nf-core
workDir : /workspaces/training/hello-nf-core/work
projectDir : /workspaces/.nextflow/assets/nf-core/demo
userName : root
profile : docker,test
configFiles : /workspaces/.nextflow/assets/nf-core/demo/nextflow.config
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
executor > local (7)
[ff/a6976b] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) | 3 of 3 ✔
[39/731ab7] NFCORE_DEMO:DEMO:SEQTK_TRIM (SAMPLE3_SE) | 3 of 3 ✔
[7c/78d96e] NFCORE_DEMO:DEMO:MULTIQC | 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
Wenn deine Ausgabe damit übereinstimmt, Glückwunsch! Du hast gerade deine erste nf-core Pipeline ausgeführt.
Du wirst bemerken, dass es viel mehr Konsolenausgabe gibt als wenn du eine einfache Nextflow-Pipeline ausführst. Es gibt einen Header, der eine Zusammenfassung der Pipeline-Version, Eingaben und Ausgaben sowie einige Konfigurationselemente enthält.
Hinweis
Deine Ausgabe wird unterschiedliche Zeitstempel, Ausführungsnamen und Dateipfade zeigen, aber die Gesamtstruktur und Prozessausführung sollte ähnlich sein.
Beachte die Zeile am Anfang der Ausgabe:
Diese zeigt dir, welche Revision der Pipeline verwendet wurde.
Da wir keine Version angegeben haben, hat Nextflow den neuesten Commit auf master verwendet.
Für reproduzierbare Ausführungen solltest du eine bestimmte Version mit dem Flag -r festlegen:
So wird sichergestellt, dass jedes Mal derselbe Pipeline-Code verwendet wird, unabhängig von neuen Commits oder Releases.
In diesem Training lassen wir -r der Einfachheit halber weg, aber in der Produktion solltest du es immer angeben.
Weiter zur Ausführungsausgabe, schauen wir uns die Zeilen an, die uns sagen, welche Prozesse ausgeführt wurden:
executor > local (7)
[ff/a6976b] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) | 3 of 3 ✔
[39/731ab7] NFCORE_DEMO:DEMO:SEQTK_TRIM (SAMPLE3_SE) | 3 of 3 ✔
[7c/78d96e] NFCORE_DEMO:DEMO:MULTIQC | 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
Dies sagt uns, dass drei Prozesse ausgeführt wurden, die den drei Tools entsprechen, die auf der Pipeline-Dokumentationsseite auf der nf-core-Website gezeigt werden: FASTQC, SEQTK_TRIM und MULTIQC.
Die vollständigen Prozessnamen wie hier gezeigt, wie NFCORE_DEMO:DEMO:MULTIQC, sind länger als das, was du im einführenden Hello Nextflow Material gesehen haben könntest.
Diese enthalten die Namen ihrer übergeordneten Workflows und spiegeln die Modularität des Pipeline-Codes wider.
Wir werden in Teil 2 dieses Kurses näher darauf eingehen.
2.3. Die Ausgaben der Pipeline untersuchen¶
Schauen wir uns schließlich das demo-results-Verzeichnis an, das von der Pipeline produziert wurde.
Verzeichnisinhalt
demo-results
├── fastqc
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── fq
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── multiqc
│ ├── multiqc_data
│ ├── multiqc_plots
│ └── multiqc_report.html
└── pipeline_info
├── execution_report_2025-11-21_04-57-41.html
├── execution_timeline_2025-11-21_04-57-41.html
├── execution_trace_2025-11-21_04-57-41.txt
├── nf_core_demo_software_mqc_versions.yml
├── params_2025-11-21_04-57-46.json
└── pipeline_dag_2025-11-21_04-57-41.html
Das mag wie viel erscheinen.
Um mehr über die Ausgaben der nf-core/demo Pipeline zu erfahren, schau dir ihre Dokumentationsseite an.
In diesem Stadium ist wichtig zu beobachten, dass die Ergebnisse nach Modul organisiert sind, und es gibt zusätzlich ein Verzeichnis namens pipeline_info, das verschiedene mit Zeitstempeln versehene Berichte über die Pipeline-Ausführung enthält.
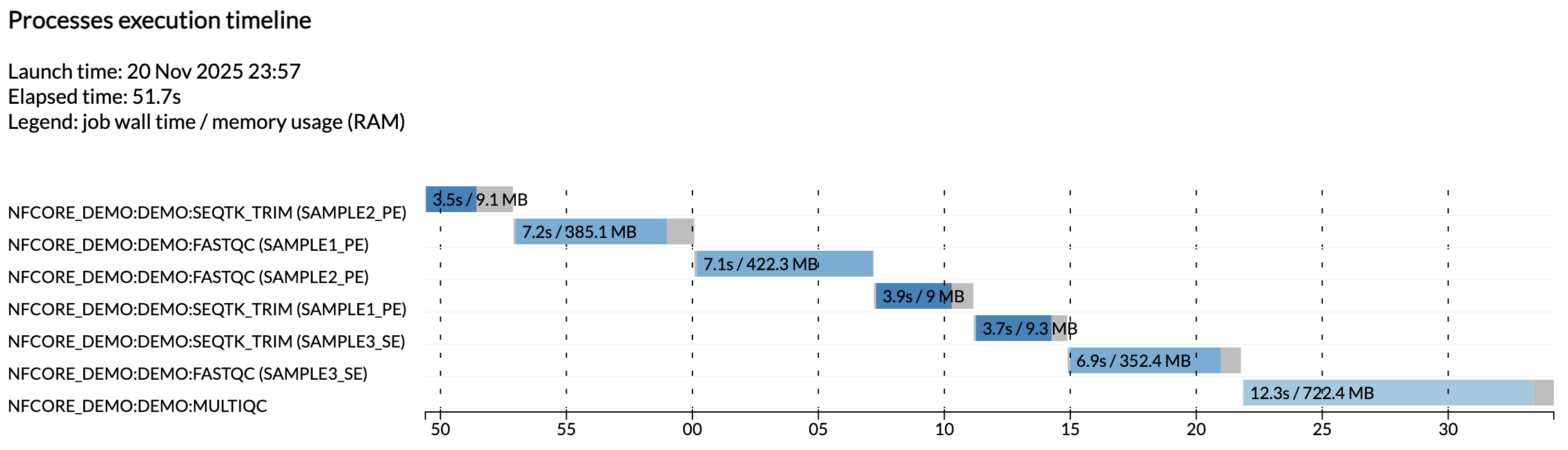
Zum Beispiel zeigt dir die Datei execution_timeline_*, welche Prozesse ausgeführt wurden, in welcher Reihenfolge und wie lange sie zur Ausführung benötigten:
Hinweis
Hier wurden die Aufgaben nicht parallel ausgeführt, weil wir auf einer minimalistischen Maschine in Github Codespaces laufen. Um diese parallel laufen zu sehen, versuche die CPU-Zuweisung deines Codespace und die Ressourcenlimits in der Testkonfiguration zu erhöhen.
Diese Berichte werden automatisch für alle nf-core Pipelines generiert.
Fazit¶
Du weißt, wie man eine nf-core Pipeline mit ihrem integrierten Testprofil ausführt und wo man ihre Ausgaben findet.
Wie geht es weiter?¶
Lerne, wie du die Pipeline konfigurierst, um ihre Ausführung anzupassen.
3. Die Pipeline-Ausführung konfigurieren¶
Wie in Hello Config erklärt, möchten wir in der Lage sein, zu ändern, auf welchen Daten unsere Pipeline läuft und wie sie läuft, ohne den Pipeline-Code selbst zu ändern. Dazu unterstützt Nextflow mehrere Möglichkeiten zur Steuerung der Pipeline-Konfiguration, was etwas überwältigend sein kann.
Das nf-core-Projekt legt Konventionen für die Organisation von Konfigurationselementen fest und unterscheidet auf oberster Ebene zwei Arten von Konfiguration: Pipeline-Parameter und Konfiguration im engeren Sinne.
- Pipeline-Parameter (über das
params-System gesetzt) umfassen typischerweise Dinge wie Eingabedateien, Tool-Verhaltens-Flags und Analyseparameter. - Konfiguration im engeren Sinne bezieht sich auf die Logistik, wie die Pipeline ausgeführt wird, d.h. den Executor, die Zuweisung von Rechenressourcen und so weiter.
Fangen wir mit den Pipeline-Parametern an, dann schauen wir uns die Konfiguration im engeren Sinne an.
3.1. Pipeline-Parameter¶
Für alle nf-core Pipelines kannst du eine vollständige Liste der Pipeline-Parameter direkt über die Befehlszeile abrufen, indem du das Flag --help verwendest, das selbst ein Pipeline-Parameter ist.
3.1.1. Die Parameterliste mit --help abrufen¶
Führe den Hilfe-Befehl für die Demo-Pipeline aus:
Befehlsausgabe
N E X T F L O W ~ version 25.10.4
Launching `https://github.com/nf-core/demo` [run_name] DSL2 - revision: 45904cb9d1 [master]
----------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.1.0
----------------------------------------------------
Typical pipeline command:
nextflow run nf-core/demo -profile <docker/singularity/.../institute> --input samplesheet.csv --outdir <OUTDIR>
Input/output options
--input [string] Path to a metadata file containing information about the samples in the experiment.
--outdir [string] The output directory where the results will be saved. You have to use absolute paths to storage on Cloud infrastructure.
--email [string] Email address for completion summary.
--multiqc_title [string] MultiQC report title. Printed as page header, used for filename if not otherwise specified.
Reference genome options
--genome [string] Name of iGenomes reference.
--fasta [string] Path to FASTA genome file.
Process skipping options
--skip_trim [boolean] Skip trimming fastq files with seqtk
Generic options
--multiqc_methods_description [string] Custom MultiQC yaml file containing HTML including a methods description.
--help [boolean, string] Display the help message.
--help_full [boolean] Display the full detailed help message.
--show_hidden [boolean] Display hidden parameters in the help message (only works when --help or --help_full are provided).
!! Hiding 20 param(s), use the `--show_hidden` parameter to show them !!
----------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
Wie du siehst, gruppiert die Ausgabe Parameter in Kategorien (Input/output options, Reference genome options, etc.) mit Typen und Beschreibungen für jeden.
Diese Kategorisierung wird durch eine Schema-Datei bestimmt, die weiter unten behandelt wird.
Bei einfachen Nextflow-Pipelines funktioniert --help nur, wenn der/die Entwickler*in es manuell implementiert hat.
Tipp
Verwende --help --show_hidden, um zusätzliche Parameter zu sehen, die standardmäßig ausgeblendet sind, wie --publish_dir_mode oder --monochrome_logs.
3.1.2. Parameterwerte setzen¶
Wie in Hello Config beschrieben, kannst du Parameterwerte über die Befehlszeile mit --param_name setzen oder eine Reihe von Parametern in einer YAML-Datei sammeln und sie mit -params-file übergeben.
Beide Ansätze funktionieren bei nf-core Pipelines genauso.
Um zum Beispiel den Trimming-Schritt zu überspringen:
Befehlsausgabe
Der Prozess SEQTK_TRIM erscheint nicht mehr in der Ausgabe.
Info
Obwohl es technisch möglich ist, Pipeline-Parameter in einer benutzerdefinierten Konfigurationsdatei zu setzen, die mit -c übergeben wird, überschreibt dies möglicherweise nicht die bereits in der eigenen nextflow.config der Pipeline gesetzten Standardwerte, abhängig von Nextflows Konfigurationsprioritätsregeln.
Die Verwendung von --param_name auf der Befehlszeile oder -params-file ist zuverlässiger, da diese immer Vorrang haben.
Als Faustregel gilt: Wenn es in der --help-Ausgabe erscheint, setze es über die Befehlszeile oder eine Params-Datei statt über eine Konfigurationsdatei.
3.1.3. Parametervalidierung¶
Interessant: Der Befehl --help funktioniert für alle nf-core Pipelines, weil das nf-core-Projekt von Entwickler*innen verlangt, alle Pipeline-Parameter formal in einer JSON-Schema-Datei (nextflow_schema.json) zu definieren.
Dieses Schema erfasst den Typ, die Beschreibung, den Standardwert und die Gruppierung jedes Parameters.
Neben der Bereitstellung der --help-Ausgabe ermöglicht die Schema-Datei auch eine automatisierte Validierung beim Start.
Das bedeutet, dass Nextflow prüfen kann, ob jeder Parameter, den du übergibst, existiert und einen geeigneten Wert erhalten hat (vom richtigen Typ, innerhalb des erlaubten Wertebereichs usw.).
Wir behandeln dies ausführlicher in Teil 5: Eingabevalidierung, aber du kannst es bereits in Aktion sehen, indem du der Demo-Pipeline ungültige Parametereingaben gibst.
3.1.3.1. Nicht erkannte Parameter¶
Versuche, einen Parameter zu übergeben, der nicht existiert:
Die Konsolenausgabe enthält eine Warnung:
Die Pipeline läuft weiterhin, aber die Warnung macht dich sofort darauf aufmerksam, dass --foobar kein erkannter Parameter ist.
Das erkennt Tippfehler wie --outDir statt --outdir, bevor du Rechenzeit damit verschwendest, dich zu fragen, warum die Ausgabe an den falschen Ort gegangen ist.
3.1.3.2. Ungültige Parameterwerte¶
Die Validierung prüft auch Parameterwerte.
Der Parameter --skip_trim ist ein boolean Flag, daher führt die Übergabe eines String-Werts dazu, dass die Pipeline sofort fehlschlägt:
ERROR ~ Validation of pipeline parameters failed!
The following invalid input values have been detected:
* --skip_trim (yes): Value is [string] but should be [boolean]
Die Pipeline stoppt, bevor irgendwelche Prozesse ausgeführt werden, und bewahrt dich so vor einer fehlgeschlagenen oder falschen Ausführung.
Boolean-Parameter sollten als Flags (--skip_trim) ohne Wert übergeben werden, oder in einer Params-Datei auf true/false gesetzt werden.
3.1.4. Eingabevalidierung¶
Dieselbe Validierungslogik kann auch verwendet werden, um die Gültigkeit von Eingabedateien zu prüfen. Wenn eine Pipeline beispielsweise ein Samplesheet als Hauptdateneingabe erwartet (was bei vielen, wenn nicht den meisten nf-core Pipelines der Fall ist), kann der/die Entwickler*in ein Eingabeschema (getrennt vom Parameterschema) bereitstellen, das beschreibt, wie die Eingabedatei strukturiert sein soll.
Zur Laufzeit kann Nextflow dann prüfen, ob die bereitgestellte Eingabedatei gültig ist.
Wir behandeln dies ebenfalls ausführlicher in Teil 5: Eingabevalidierung, aber du kannst es bereits in Aktion sehen, indem du der Demo-Pipeline ein ungültiges Eingabe-Samplesheet gibst.
Die nf-core/demo Pipeline erwartet eine CSV-Datei mit den Spalten sample, fastq_1 und fastq_2.
Dies ist in einer Schema-Datei (assets/schema_input.json) definiert, die die erwartete Struktur, Spaltentypen und Einschränkungen festlegt.
assets/schema_input.json
{
"$schema": "https://json-schema.org/draft/2020-12/schema",
"$id": "https://raw.githubusercontent.com/nf-core/demo/master/assets/schema_input.json",
"title": "nf-core/demo pipeline - params.input schema",
"description": "Schema for the file provided with params.input",
"type": "array",
"items": {
"type": "object",
"properties": {
"sample": {
"type": "string",
"pattern": "^\\S+$",
"errorMessage": "Sample name must be provided and cannot contain spaces",
"meta": ["id"]
},
"fastq_1": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 1 must be provided, cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
},
"fastq_2": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 2 cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
}
},
"required": ["sample", "fastq_1"]
}
}
Das Schema legt fest, dass sample und fastq_1 erforderlich sind, während fastq_2 optional ist (unterstützt sowohl Paired-End- als auch Single-End-Daten).
Dateipfade werden auf Existenz und Erweiterungsmuster validiert.
3.1.4.1. Ein ungültiges Samplesheet erstellen¶
Erstelle ein Samplesheet mit einer fehlenden Spalte und einem nicht existierenden Dateipfad:
Diesem Samplesheet fehlt die erforderliche Spalte fastq_1 und es enthält einen nicht existierenden Dateipfad in fastq_2.
Beide Probleme werden im nächsten Schritt Validierungsfehler erzeugen.
3.1.4.2. Die Demo-Pipeline mit dem ungültigen Samplesheet ausführen¶
Führe die Demo-Pipeline mit malformed_samplesheet.csv als Eingabe aus.
nextflow run nf-core/demo -profile docker,test --outdir demo-results --input malformed_samplesheet.csv
ERROR ~ Validation of pipeline parameters failed!
The following invalid input values have been detected:
* --input (malformed_samplesheet.csv): Validation of file failed:
-> Entry 1: Error for field 'fastq_2' (/not/a/real/file.fastq.gz): the file or directory
'/not/a/real/file.fastq.gz' does not exist (FastQ file for reads 2 cannot contain spaces
and must have extension '.fq.gz' or '.fastq.gz')
-> Entry 1: Missing required field(s): fastq_1
Wie du siehst, schlägt die Pipeline sofort fehl und meldet alle Validierungsfehler auf einmal. nf-schema stoppt nicht beim ersten Fehler – es sammelt alle Probleme und listet sie zusammen auf, sodass du alles auf einmal beheben kannst, anstatt Probleme einzeln zu entdecken.
Jeder Fehler identifiziert den genauen Eintrag und das Feld, das das Problem verursacht hat, sodass du dein Samplesheet korrigieren und die Pipeline dann mit der Gewissheit neu starten kannst, dass sie nicht an einem späteren Punkt fehlschlägt, wenn Nextflow tatsächlich auf den Dateipfad zugreift.
Für Entwickler*innen wird all dies ausführlicher in Teil 5 dieses Kurses behandelt.
3.2. Konfiguration¶
Konfiguration im engeren Sinne steuert, wie die Pipeline läuft: Ressourcenzuweisung, Tool-spezifische Argumente, wo Jobs ausgeführt werden und welches Software-Packaging-System verwendet wird.
nf-core Pipelines enthalten Standardkonfiguration in nextflow.config und dem conf/-Verzeichnis.
Bevor du etwas überschreibst, ist es hilfreich zu wissen, wo die Standardwerte liegen.
Du hast in Abschnitt 2.1 bereits gesehen, dass der Pipeline-Quellcode in $NXF_HOME/assets liegt.
Liste die Konfigurationsdateien auf, um zu sehen, was verfügbar ist:
Die wichtigsten Konfigurationsdateien sind:
conf/base.config: Definiert Ressourcen-Labels (process_low,process_medium,process_high), die Prozessen CPUs, Arbeitsspeicher und Zeit zuweisen. Wenn du siehst, dass ein Prozess mehr Ressourcen als erwartet verwendet, kommen diese Standardwerte von hier.conf/modules.config: Setzt prozessspezifische Tool-Argumente (ext.args) und Einstellungen für die Ausgabeveröffentlichung (publishDir). Öffne diese Datei, um zu sehen, welche Argumente jedes Tool standardmäßig erhält.conf/test.config: Das Testprofil, das du in Abschnitt 2.1 verwendet hast. Es begrenzt Ressourcen überresourceLimitsund setzt ein Test-Samplesheet. Wird mit-profile testaktiviert. Es gibt auch eineconf/test_full.configfür die Ausführung mit einem vollständigen Testdatensatz, nützlich für Benchmarking.
Die zentrale nextflow.config lädt all das oben Genannte und setzt die entsprechenden Standardwerte für alles.
Wenn du Einstellungen aus diesen Dateien ändern möchtest, ändere keine dieser Dateien direkt.
Erstelle stattdessen deine eigene Konfigurationsdatei und übergebe sie mit -c.
Die von dir angegebenen Werte überschreiben die in diesen anderen Dateien gesetzten Standardwerte.
Lass uns einige Übungen durchgehen, um das in der Praxis zu tun.
3.2.1. Ressourcenzuweisung für einen Prozess ändern¶
Die Demo-Pipeline weist Ressourcen über Labels zu, die in base.config definiert sind.
Zum Beispiel verwendet FASTQC das Label process_medium, das 6 CPUs und 36 GB Arbeitsspeicher zuweist.
Das Testprofil begrenzt Ressourcen über resourceLimits, aber du kannst auch Ressourcen für bestimmte Prozesse überschreiben.
Erstelle eine Datei namens custom.config:
Führe die Pipeline mit deiner benutzerdefinierten Konfiguration aus:
Befehlsausgabe
Das Flag -c fügt deine Konfiguration zusätzlich zur integrierten Konfiguration der Pipeline hinzu.
3.2.2. Tool-Argumentwerte mit ext.args setzen¶
Viele Befehlszeilen-Tools haben Argumente, die nicht erforderlich sind und daher nicht als Pipeline-Parameter eingerichtet werden, es sei denn, sie werden sehr häufig verwendet.
Für diese Tool-Argumente verwenden nf-core Module eine Nextflow-Konvention namens ext.args, um Argumente über eine Konfigurationsdatei an das zugrunde liegende Tool zu übergeben.
Lass uns zum Beispiel ein Trimming-Argument zum SEQTK_TRIM-Modul mit ext.args hinzufügen.
3.2.2.1. Die benutzerdefinierte Konfiguration aktualisieren¶
Aktualisiere deine custom.config:
| custom.config | |
|---|---|
Dies weist seqtk trimfq an, zusätzlich zum Qualitäts-Trimming 5 Basen vom Anfang jedes Reads zu trimmen.
3.2.2.2. Die Pipeline ausführen¶
Führe die Pipeline erneut mit dieser Konfiguration aus, um den Effekt zu sehen:
Befehlsausgabe
Um zu überprüfen, ob das Argument angewendet wurde, suche den Hash des SEQTK_TRIM work-Verzeichnisses aus der Ausführungsausgabe (z.B. work/ab/cd1234...) und prüfe die Datei .command.sh darin:
Befehlsausgabe
Du solltest -b 5 im seqtk trimfq-Befehl sehen, was bestätigt, dass deine ext.args-Überschreibung wirksam war.
3.2.2.3. Standardwerte überschreiben¶
Einige Module haben ext.args bereits standardmäßig gesetzt.
Zum Beispiel ist das FASTQC-Modul standardmäßig mit ext.args = '--quiet' konfiguriert (definiert in conf/modules.config).
| conf/modules.config | |
|---|---|
Wenn du einen Wert für ext.args über eine benutzerdefinierte Konfigurationsdatei angibst, ersetzt dieser Wert den für diesen Prozess gesetzten Standard vollständig.
Wenn der Standard zum Beispiel '--quiet' war und du ext.args = '--kmers 8' setzt, wird das Flag --quiet nicht mehr angewendet.
Um beides beizubehalten, setze ext.args = '--quiet --kmers 8'.
Das bedeutet, dass du dafür verantwortlich bist, die Standardkonfiguration der Tools zu prüfen, denen du Argumentwerte mit ext.args übergeben möchtest.
Fazit¶
Du weißt, wie du Hilfe von einer nf-core Pipeline erhältst, Parameter setzt und verstehst, wie sie validiert werden, und wie du die Konfiguration über Konfigurationsdateien anpasst.
Wie geht es weiter?¶
Mach eine Pause! Wenn du bereit bist, gehe zu Teil 2 über, wo du deine eigene nf-core-kompatible Pipeline von Grund auf erstellen wirst.