Part 8: Hello nf-core¶
nf-core is a community effort to develop and maintain a curated set of analysis pipelines built using Nextflow.
![]()
nf-core provides a standardized set of best practices, guidelines, and templates for building and sharing scientific pipelines. These pipelines are designed to be modular, scalable, and portable, allowing researchers to easily adapt and execute them using their own data and compute resources.
One of the key benefits of nf-core is that it promotes open development, testing, and peer review, ensuring that the pipelines are robust, well-documented, and validated against real-world datasets. This helps to increase the reliability and reproducibility of scientific analyses and ultimately enables researchers to accelerate their scientific discoveries.
nf-core is published in Nature Biotechnology: Nat Biotechnol 38, 276–278 (2020). Nature Biotechnology. An updated preprint is available at bioRxiv.
nf-core pipelines and other components¶
The nf-core collection currently offers over 100 pipelines in various stages of development, 72 subworkflows and over 1300 modules that you can use to build your own pipelines.
Each released pipeline has a dedicated page that includes 6 documentation sections:
- Introduction: An introduction and overview of the pipeline
- Usage: Descriptions of how to execute the pipeline
- Parameters: Grouped pipeline parameters with descriptions
- Output: Descriptions and examples of the expected output files
- Results: Example output files generated from the full test dataset
- Releases & Statistics: Pipeline version history and statistics
You should read the pipeline documentation carefully to understand what a given pipeline does and how it can be configured before attempting to run it.
Pulling an nf-core pipeline¶
One really cool aspect of how Nextflow manages pipelines is that you can pull a pipeline from a GitHub repository without cloning the repository. This is really convenient if you just want to run a pipeline without modifying the code.
So if you want to try out an nf-core pipeline with minimal effort, you can start by pulling it using the nextflow pull command.
Tip
You can run this from anywhere, but if you feel like being consistent with previous exercises, you can create a nf-core-demo directory under hello-nextflow. If you were working through Part 7 (Hello nf-test) before this, you may need to go up one level first.
Whenever you're ready, run the command:
Nextflow will pull the pipeline's default GitHub branch.
For nf-core pipelines with a stable release, that will be the master branch.
You select a specific branch with -r; we'll cover that later.
Checking nf-core/demo ...
downloaded from https://github.com/nf-core/demo.git - revision: 04060b4644 [master]
To be clear, you can do this with any Nextflow pipeline that is appropriately set up in GitHub, not just nf-core pipelines. However nf-core is the largest open curated collection of Nextflow pipelines.
Tip
Pulled pipelines are stored in a hidden assets folder. By default, this folder is $HOME/.nextflow/assets, but in this training environment the folder has been set to $NXF_HOME/assets:
So you don't actually see them listed in your working directory.
However, you can view a list of your cached pipelines using the nextflow list command:
Now that we've got the pipeline pulled, we can try running it!
Trying out an nf-core pipeline with the test profile¶
Conveniently, every nf-core pipeline comes with a test profile.
This is a minimal set of configuration settings for the pipeline to run using a small test dataset that is hosted on the nf-core/test-datasets repository. It's a great way to try out a pipeline at small scale.
The test profile for nf-core/demo is shown below:
This tells us that the nf-core/demo test profile already specifies the input parameter, so you don't have to provide any input yourself.
However, the outdir parameter is not included in the test profile, so you have to add it to the execution command using the --outdir flag.
Here, we're also going to specify -profile docker, which by nf-core convention enables the use of Docker.
Lets' try it!
Changing Nextflow version
Depending on the Nextflow version you have installed, this command might fail due to a version mismatch.
If that happens, you can temporarily run the pipeline with a different version than you have installed by adding NXF_VER=version to the start of your command as shown below:
Here's the console output from the pipeline:
N E X T F L O W ~ version 24.09.2-edge
Launching `https://github.com/nf-core/demo` [naughty_bell] DSL2 - revision: 04060b4644 [master]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.0.1
------------------------------------------------------
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : results
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Core Nextflow options
revision : master
runName : naughty_bell
containerEngine : docker
launchDir : /workspace/gitpod/hello-nextflow
workDir : /workspace/gitpod/hello-nextflow/work
projectDir : /home/gitpod/.nextflow/assets/nf-core/demo
userName : gitpod
profile : docker,test
configFiles :
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
executor > local (7)
[0a/e694d8] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) [100%] 3 of 3 ✔
[85/4198c1] NFCORE_DEMO:DEMO:SEQTK_TRIM (SAMPLE1_PE) [100%] 3 of 3 ✔
[d8/fe153e] NFCORE_DEMO:DEMO:MULTIQC [100%] 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
Completed at: 28-Oct-2024 03:24:58
Duration : 1m 13s
CPU hours : (a few seconds)
Succeeded : 7
Isn't that neat?
You can also explore the results directory produced by the pipeline.
results
├── fastqc
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── fq
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── multiqc
│ ├── multiqc_data
│ ├── multiqc_plots
│ └── multiqc_report.html
└── pipeline_info
├── execution_report_2024-10-28_03-23-44.html
├── execution_timeline_2024-10-28_03-23-44.html
├── execution_trace_2024-10-28_03-14-32.txt
├── execution_trace_2024-10-28_03-19-33.txt
├── execution_trace_2024-10-28_03-20-57.txt
├── execution_trace_2024-10-28_03-22-39.txt
├── execution_trace_2024-10-28_03-23-44.txt
├── nf_core_pipeline_software_mqc_versions.yml
├── params_2024-10-28_03-23-49.json
└── pipeline_dag_2024-10-28_03-23-44.html
If you're curious about what that all means, check out the nf-core/demo pipeline documentation page!
And that's all you need to know for now. Congratulations! You have now run your first nf-core pipeline.
Takeaway¶
You have a general idea of what nf-core offers and you know how to run an nf-core pipeline using its built-in test profile.
What's next?¶
Celebrate and take another break! Next, we'll show you how to use nf-core tooling to build your own pipeline.
Create a basic pipeline from template¶
We will now start developing our own nf-core style pipeline. The nf-core community provides a command line tool with helper functions to use and develop pipelines. We have pre-installed nf-core tools, and here, we will use them to create and develop a new pipeline.
View all of the tooling using the nf-core --help argument.
Creating your pipeline¶
Before we start, let's navigate into the hello-nf-core directory:
Open a new window in VSCode
If you are working with VS Code you can open a new window to reduce visual clutter:
Let's start by creating a new pipeline with the nf-core pipelines create command:
All nf-core pipelines are based on a common template, a standardized pipeline skeleton that can be used to streamline development with shared features and components.
The nf-core pipelines create command creates a new pipeline using the nf-core base template with a pipeline name, description, and author. It is the first and most important step for creating a pipeline that will integrate with the wider Nextflow ecosystem.
Running this command will open a Text User Interface (TUI) for pipeline creation.
Template features can be flexibly included or excluded at the time of creation, follow these steps create your first pipeline using the nf-core pipelines create TUI:
- Run the
nf-core pipelines createcommand - Select Let's go! on the welcome screen
- Select Custom on the Choose pipeline type screen
-
Enter your pipeline details, replacing < YOUR NAME > with your own name, then select Next
- GitHub organisation: myorg
- Workflow name: myfirstpipeline
- A short description of your pipeline: My first pipeline
- Name of the main author / authors: < YOUR NAME >
-
On the Template features screen, turn off:
Use a GitHub repositoryAdd Github CI testsUse reference genomesAdd Github badgesInclude citationsInclude a gitpod environmentInclude GitHub CodespacesUse fastqcAdd a changelogSupport Microsoft Teams notificationsSupport Slack notifications
-
Select Finish on the Final details screen
- Wait for the pipeline to be created, then select Continue
- Select Finish without creating a repo on the Create GitHub repository screen
- Select Close on the HowTo create a GitHub repository page
If run successfully, you will see a new folder in your current directory named myorg-myfirstpipeline.
Testing your pipeline¶
Let's try to run our new pipeline:
The pipeline should run successfully!
Here's the console output from the pipeline:
Launching `./main.nf` [marvelous_saha] DSL2 - revision: a633aedb88
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : results
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Core Nextflow options
runName : marvelous_saha
containerEngine : docker
launchDir : /workspace/gitpod/hello-nextflow/hello-nf-core/myorg-myfirstpipeline
workDir : /workspace/gitpod/hello-nextflow/hello-nf-core/myorg-myfirstpipeline/work
projectDir : /workspace/gitpod/hello-nextflow/hello-nf-core/myorg-myfirstpipeline
userName : gitpod
profile : docker,test
configFiles :
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
executor > local (1)
[ba/579181] process > MYORG_MYFIRSTPIPELINE:MYFIRSTPIPELINE:MULTIQC [100%] 1 of 1 ✔
-[myorg/myfirstpipeline] Pipeline completed successfully-
Let's dissect what we are seeing.
The nf-core pipeline template is a working pipeline and comes preconfigured with some modules. Here, we only run MultiQC
At the top, you see all parameters displayed that differ from the pipeline defaults. Most of these are default or were set by applying the test profile.
Additionally we used the docker profile to use docker for software packaging. nf-core provides this as a profile for convenience to enable the docker feature but we could do it with configuration as we did with the earlier module.
Template tour¶
The nf-core pipeline template comes packed with a lot of files and folders. While creating the pipeline, we selected a subset of the nf-core features. The features we selected are now included as files and directories in our repository.
While the template may feel overwhelming, a complete understanding isn't required to start developing your pipeline. Let's look at the important places that we need to touch during pipeline development.
Workflows, subworkflows, and modules¶
The nf-core pipeline template has a main.nf script that calls myfirstpipeline.nf from the workflows folder. The myfirstpipeline.nf file inside the workflows folder is the central pipeline file that is used to bring everything else together.
Instead of having one large monolithic pipeline script, it's broken up into smaller script components, namely, modules and subworkflows:
- Modules: Wrappers around a single process
- Subworkflows: Two or more modules that are packaged together as a mini workflow
Within your pipeline repository, modules and subworkflows are stored within local and nf-core folders. The nf-core folder is for components that have come from the nf-core GitHub repository while the local folder is for components that have been developed independently (usually things very specific to a pipeline):
modules/
├── local
│ └── <toolname>.nf
│ .
│
└── nf-core
├── <tool name>
│ ├── environment.yml
│ ├── main.nf
│ ├── meta.yml
│ └── tests
│ ├── main.nf.test
│ ├── main.nf.test.snap
│ └── tags.yml
.
Modules from nf-core follow a similar structure and contain a small number of additional files for testing using nf-test and documentation about the module.
Note
Some nf-core modules are also split into command specific directories:
Note
The nf-core template does not come with a local modules folder by default.
Configuration files¶
The nf-core pipeline template utilizes Nextflow's flexible customization options and has a series of configuration files throughout the template.
In the template, the nextflow.config file is a central configuration file and is used to set default values for parameters and other configuration options. The majority of these configuration options are applied by default while others (e.g., software dependency profiles) are included as optional profiles.
There are several configuration files that are stored in the conf folder and are added to the configuration by default or optionally as profiles:
base.config: A 'blank slate' config file, appropriate for general use on most high-performance computing environments. This defines broad bins of resource usage, for example, which are convenient to apply to modules.modules.config: Additional module directives and arguments.test.config: A profile to run the pipeline with minimal test data.test_full.config: A profile to run the pipeline with a full-sized test dataset.
nextflow_schema.json¶
The nextflow_schema.json is a file used to store parameter related information including type, description and help text in a machine readable format. The schema is used for various purposes, including automated parameter validation, help text generation, and interactive parameter form rendering in UI interfaces.
assets/schema_input.json¶
The schema_input.json is a file used to define the input samplesheet structure. Each column can have a type, pattern, description and help text in a machine readable format. The schema is used for various purposes, including automated validation, and providing helpful error messages.
Takeaway¶
You have an example pipeline, and learned about important template files.
What's next?¶
Congratulations! In the next step, we will check the input data.
Check the input data¶
Above, we said that the test profile comes with small test files that are stored in the nf-core. Let's check what type of files we are dealing with to plan our expansion. Remember that we can inspect any channel content using the view operator:
| workflows/myfirstpipeline.nf | |
|---|---|
and the run command:
The output should look like the below. We see that we have FASTQ files as input and each set of files is accompanied by some metadata: the id and whether or not they are single end:
[['id':'SAMPLE1_PE', 'single_end':false], [/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz, /nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R2.fastq.gz]]
[['id':'SAMPLE2_PE', 'single_end':false], [/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz, /nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R2.fastq.gz]]
[['id':'SAMPLE3_SE', 'single_end':true], [/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz, /nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz]]
You can comment the view statement for now. We will use later during this training to inspect the channel content again.
Takeaway¶
You have learned how input data is supplied via a samplesheet.
What's next?¶
In the next step we will start changing the code and add new tools to the pipeline.
Add an nf-core module¶
nf-core provides a large library of modules and subworkflows: pre-made nextflow wrappers around tools that can be installed into nextflow pipelines. They are designed to be flexible but may require additional configuration to suit different use cases.
Currently, there are more than 1300 nf-core modules and 60 nf-core subworkflows (November 2024) available. Modules and subworkflows can be listed, installed, updated, removed, and patched using nf-core tooling.
While you could develop a module for this tool independently, you can save a lot of time and effort by leveraging nf-core modules and subworkflows.
Let's see which modules are available:
This command lists all currently available modules, > 1300. An easier way to find them is to go to the nf-core website and visit the modules subpage https://nf-co.re/modules. Here you can search for modules by name or tags, find documentation for each module, and see which nf-core pipeline are using the module:
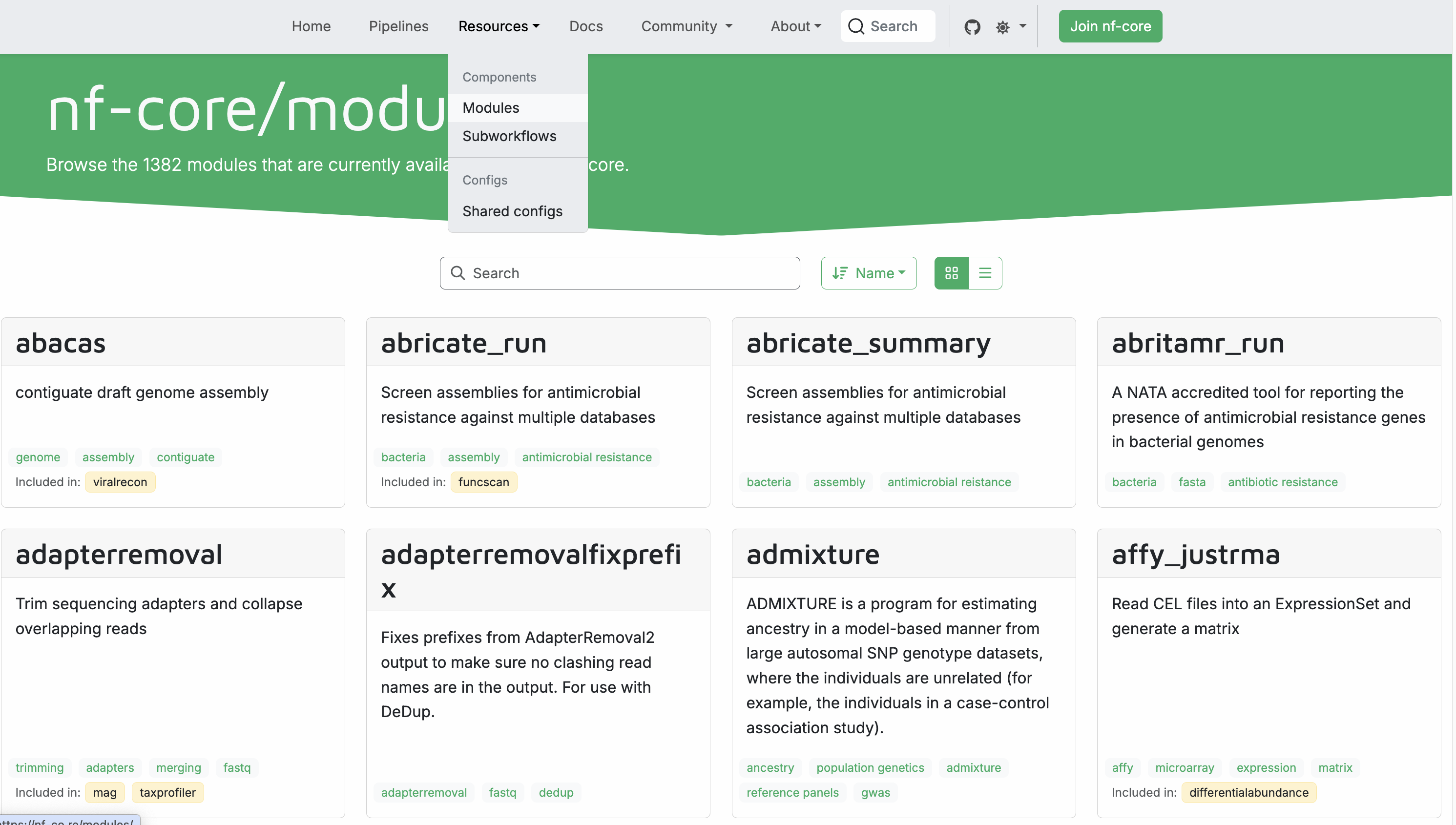
Install an nf-core module¶
Now let's add another tool to the pipeline.
Seqtk is a fast and lightweight tool for processing sequences in the FASTA or FASTQ format. Here, you will use the seqtk trim command to trim FASTQ files.
In your pipeline, you will add a new step that will take FASTQ files from the sample sheet as inputs and will produce trimmed fastq files that can be used as an input for other tools and version information about the seqtk tools to mix into the inputs for the MultiQC process.
The nf-core modules install command can be used to install the seqtk/trim module directly from the nf-core repository:
Warning
You need to be in the myorg-myfirstpipeline directory when executing nf-core modules install
You can follow the prompts to find and install the module you are interested in:
Once selected, the tooling will install the module in the modules/nf-core/ folder and suggest code that you can add to your main workflow file (workflows/myfirstpipeline.nf).
INFO Installing 'seqtk/trim'
INFO Use the following statement to include this module:
include { SEQTK_TRIM } from '../modules/nf-core/seqtk/trim/main'
To enable reporting and reproducibility, modules and subworkflows from the nf-core repository are tracked using hashes in the modules.json file. When modules are installed or removed using the nf-core tooling the modules.json file will be automatically updated.
When you open the modules.json, you will see an entry for each module that is currently installed from the nf-core modules repository. You can open the file with the VS Code user interface by clicking on it in myorg-myfirstpipeline/modules.json:
"nf-core": {
"multiqc": {
"branch": "master",
"git_sha": "cf17ca47590cc578dfb47db1c2a44ef86f89976d",
"installed_by": ["modules"]
},
"seqtk/trim": {
"branch": "master",
"git_sha": "666652151335353eef2fcd58880bcef5bc2928e1",
"installed_by": ["modules"]
}
}
Add the module to your pipeline¶
Although the module has been installed in your local pipeline repository, it is not yet added to your pipeline.
The suggested include statement needs to be added to your workflows/myfirstpipeline.nf file and the process call (with inputs) needs to be added to the workflow block.
| workflows/myfirstpipeline.nf | |
|---|---|
To add the SEQTK_TRIM module to your workflow you will need to check what inputs are required.
You can view the input channels for the module by opening the ./modules/nf-core/seqtk/trim/main.nf file.
Each nf-core module also has a meta.yml file which describes the inputs and outputs. This meta file is rendered on the nf-core website, or can be viewed using the nf-core modules info command:
It outputs a table with all defined inputs and outputs of the module:
╭─ Module: seqtk/trim ─────────────────────────────────────────────────────────────────────────────╮
│ Location: modules/nf-core/seqtk/trim │
│ 🔧 Tools: seqtk │
│ 📖 Description: Trim low quality bases from FastQ files │
╰───────────────────────────────────────────────────────────────────────────────────────────────────╯
╷ ╷
📥 Inputs │Description │ Pattern
╺━━━━━━━━━━━━━━┿━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┿━━━━━━━━━━━━╸
input[0] │ │
╶──────────────┼───────────────────────────────────────────────────────────────────────┼────────────╴
meta (map) │Groovy Map containing sample information e.g. [ id:'test', │
│single_end:false ] │
╶──────────────┼───────────────────────────────────────────────────────────────────────┼────────────╴
reads (file)│List of input FastQ files │*.{fastq.gz}
╵ ╵
╷ ╷
📥 Outputs │Description │ Pattern
╺━━━━━━━━━━━━━━━━━━━━━┿━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┿━━━━━━━━━━━━╸
reads │ │
╶─────────────────────┼────────────────────────────────────────────────────────────────┼────────────╴
meta (map) │Groovy Map containing sample information e.g. [ id:'test', │
│single_end:false ] │
╶─────────────────────┼────────────────────────────────────────────────────────────────┼────────────╴
*.fastq.gz (file) │Filtered FastQ files │*.{fastq.gz}
╶─────────────────────┼────────────────────────────────────────────────────────────────┼────────────╴
versions │ │
╶─────────────────────┼────────────────────────────────────────────────────────────────┼────────────╴
versions.yml (file)│File containing software versions │versions.yml
╵ ╵
Use the following statement to include this module:
include { SEQTK_TRIM } from '../modules/nf-core/seqtk/trim/main'
Using this module information you can work out what inputs are required for the SEQTK_TRIM process:
-
tuple val(meta), path(reads)- A tuple with a meta map and a list of FASTQ files
- The channel
ch_samplesheetused by theFASTQCprocess can be used as the reads input.
Only one input channel is required, and it already exists, so it can be added to your firstpipeline.nf file without any additional channel creation or modifications.
Before:
After:
| workflows/myfirstpipeline.nf | |
|---|---|
Let's test it:
Launching `./main.nf` [drunk_waddington] DSL2 - revision: a633aedb88
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : results
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Core Nextflow options
runName : drunk_waddington
containerEngine : docker
launchDir : /workspace/gitpod/hello-nextflow/hello-nf-core/myorg-myfirstpipeline
workDir : /workspace/gitpod/hello-nextflow/hello-nf-core/myorg-myfirstpipeline/work
projectDir : /workspace/gitpod/hello-nextflow/hello-nf-core/myorg-myfirstpipeline
userName : gitpod
profile : docker,test
configFiles :
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
executor > local (4)
[74/9b2e7b] process > MYORG_MYFIRSTPIPELINE:MYFIRSTPIPELINE:SEQTK_TRIM (SAMPLE2_PE) [100%] 3 of 3 ✔
[ea/5ca001] process > MYORG_MYFIRSTPIPELINE:MYFIRSTPIPELINE:MULTIQC [100%] 1 of 1 ✔
-[myorg/myfirstpipeline] Pipeline completed successfully-
Inspect results folder¶
Default nf-core configuration directs the output of each process into the <outdir>/<TOOL>. After running the previous command, you
should have a results folder that looks something like this:
results
├── multiqc
│ ├── multiqc_data
│ └── multiqc_report.html
├── pipeline_info
│ ├── execution_report_2024-11-14_12-07-43.html
│ ├── execution_report_2024-11-14_12-12-42.html
│ ├── execution_report_2024-11-14_12-13-58.html
│ ├── execution_report_2024-11-14_12-28-59.html
│ ├── execution_timeline_2024-11-14_12-07-43.html
│ ├── execution_timeline_2024-11-14_12-12-42.html
│ ├── execution_timeline_2024-11-14_12-13-58.html
│ ├── execution_timeline_2024-11-14_12-28-59.html
│ ├── execution_trace_2024-11-14_12-07-43.txt
│ ├── execution_trace_2024-11-14_12-12-42.txt
│ ├── execution_trace_2024-11-14_12-13-58.txt
│ ├── execution_trace_2024-11-14_12-28-59.txt
│ ├── params_2024-11-14_12-07-44.json
│ ├── params_2024-11-14_12-12-43.json
│ ├── params_2024-11-14_12-13-59.json
│ ├── params_2024-11-14_12-29-00.json
│ ├── pipeline_dag_2024-11-14_12-07-43.html
│ ├── pipeline_dag_2024-11-14_12-12-42.html
│ ├── pipeline_dag_2024-11-14_12-13-58.html
│ ├── pipeline_dag_2024-11-14_12-28-59.html
│ └── pipeline_software_mqc_versions.yml
└── seqtk
├── SAMPLE1_PE_sample1_R1.fastq.gz
├── SAMPLE1_PE_sample1_R2.fastq.gz
├── SAMPLE2_PE_sample2_R1.fastq.gz
├── SAMPLE2_PE_sample2_R2.fastq.gz
├── SAMPLE3_SE_sample1_R1.fastq.gz
└── SAMPLE3_SE_sample2_R1.fastq.gz
The outputs from the multiqc and seqtk modules are published in their respective subdirectories. In addition, by default,nf-core' pipelines generate a set of reports. These files are stored in thepipeline_info` subdirectory and time-stamped so that runs don't overwrite each other.
Handle modules output¶
As with the inputs, you can view the outputs for the module by opening the /modules/nf-core/seqtk/trim/main.nf file and viewing the module metadata.
| modules/nf-core/seqtk/trim/main.nf | |
|---|---|
To help with organization and readability it is beneficial to create named output channels.
For SEQTK_TRIM, the reads output could be put into a channel named ch_trimmed.
| workflows/myfirstpipeline.nf | |
|---|---|
Similarly, it is beneficial to immediately mix the tool versions into the ch_versions channel so they can be used as input for the MULTIQC process and passed to the final report.
| workflows/myfirstpipeline.nf | |
|---|---|
Note
The first operator is used to emit the first item from SEQTK_TRIM.out.versions to avoid duplication.
Add a parameter to the seqtk/trim tool¶
nf-core modules should be flexible and usable across many different pipelines. Therefore, tool parameters are typically not set in an nf-core/module. Instead, additional configuration options on how to run the tool, like its parameters or filename, can be applied to a module using the conf/modules.config file on the pipeline level. Process selectors (e.g., withName) are used to apply configuration options to modules selectively. Process selectors must be used within the process scope.
The parameters or arguments of a tool can be changed using the directive args. You can find many examples of how arguments are added to modules in nf-core pipelines, for example, the nf-core/demo modules.config file.
Add this snippet to your conf/modules.config file (using the params scope) to call the seqtk/trim tool with the argument -b 5 to trim 5 bp from the left end of each read:
Run the pipeline again and check if the new parameter is applied:
nextflow run . -profile docker,test --outdir results
[6c/34e549] process > MYORG_MYFIRSTPIPELINE:MYFIRSTPIPELINE:SEQTK_TRIM (SAMPLE1_PE) [100%] 3 of 3 ✔
[27/397ccf] process > MYORG_MYFIRSTPIPELINE:MYFIRSTPIPELINE:MULTIQC [100%] 1 of 1 ✔
Copy the hash you see in your console output (here 6c/34e549; it is different for each run). You can ls using tab-completion in your work directory to expand the complete hash.
In this folder you will find various log files. The .command.sh file contains the resolved command:
We can see, that the parameter -b 5, that we set in the modules.config is applied to the task:
#!/usr/bin/env bash
set -e # Exit if a tool returns a non-zero status/exit code
set -u # Treat unset variables and parameters as an error
set -o pipefail # Returns the status of the last command to exit with a non-zero status or zero if all successfully execute
set -C # No clobber - prevent output redirection from overwriting files.
printf "%s\n" sample1_R1.fastq.gz sample1_R2.fastq.gz | while read f;
do
seqtk \
trimfq \
-b 5 \
$f \
| gzip --no-name > SAMPLE1_PE_$(basename $f)
done
cat <<-END_VERSIONS > versions.yml
"MYORG_MYFIRSTPIPELINE:MYFIRSTPIPELINE:SEQTK_TRIM":
seqtk: $(echo $(seqtk 2>&1) | sed 's/^.*Version: //; s/ .*$//')
END_VERSIONS
Takeaway¶
You have now added a nf-core/module to your pipeline, configured it with a particular parameter, and made the output available in the workflow.
What's next?¶
In the next step we will add a pipeline parameter to allow users to skip the trimming step.
Adding parameters to your pipeline¶
Anything that a pipeline user may want to configure regularly should be made into a parameter so it can easily be overridden. nf-core defines some standards for providing parameters.
Here, as a simple example, you will add a new parameter to your pipeline that will skip the SEQTK_TRIM process.
Parameters are accessible in the pipeline script.
Default values¶
In the nf-core template the default values for parameters are set in the nextflow.config in the base repository.
Any new parameters should be added to the nextflow.config with a default value within the params scope.
Parameter names should be unique and easily identifiable.
We can a new parameter skip_trim to your nextflow.config file and set it to false.
Adding parameters to your pipeline¶
Here, an if statement that is depended on the skip_trim parameter can be used to control the execution of the SEQTK_TRIM process. An ! can be used to imply the logical "not".
Thus, if the skip_trim parameter is not true, the SEQTK_TRIM will be be executed.
| workflows/myfirstpipeline.nf | |
|---|---|
Now your if statement has been added to your main workflow file and has a default setting in your nextflow.config file, you will be able to flexibly skip the new trimming step using the skip_trim parameter.
We can now run the pipeline with the new skip_trim parameter to check it is working:
You should see that the SEQTK_TRIM process has been skipped in your execution:
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
WARN: The following invalid input values have been detected:
* --skip_trim: true
executor > local (1)
[7b/8b60a0] process > MYORG_MYFIRSTPIPELINE:MYFIRSTPIPELINE:MULTIQC [100%] 1 of 1 ✔
-[myorg/myfirstpipeline] Pipeline completed successfully-
Validate input parameters¶
When we ran the pipeline, we saw a warning message:
Parameters are validated through the nextflow_schema.json file. This file is also used by the nf-core website (for example, in nf-core/mag) to render the parameter documentation and print the pipeline help message (nextflow run . --help). If you have added parameters and they have not been documented in the nextflow_schema.json file, then the input validation does not recognize the parameter.
The nextflow_schema.json file can get very big and very complicated very quickly.
The nf-core pipelines schema build command is designed to support developers write, check, validate, and propose additions to your nextflow_schema.json file.
It will enable you to launch a web builder to edit this file in your web browser rather than trying to edit this file manually.
INFO [✓] Default parameters match schema validation
INFO [✓] Pipeline schema looks valid (found 20 params)
✨ Found 'params.skip_trim' in the pipeline config, but not in the schema. Add to pipeline schema? [y/n]: y
INFO Writing schema with 21 params: 'nextflow_schema.json'
🚀 Launch web builder for customization and editing? [y/n]: y
Using the web builder you can add add details about your new parameters.
The parameters that you have added to your pipeline will be added to the bottom of the nf-core pipelines schema build file. Some information about these parameters will be automatically filled based on the default value from your nextflow.config. You will be able to categorize your new parameters into a group, add icons, and add descriptions for each.
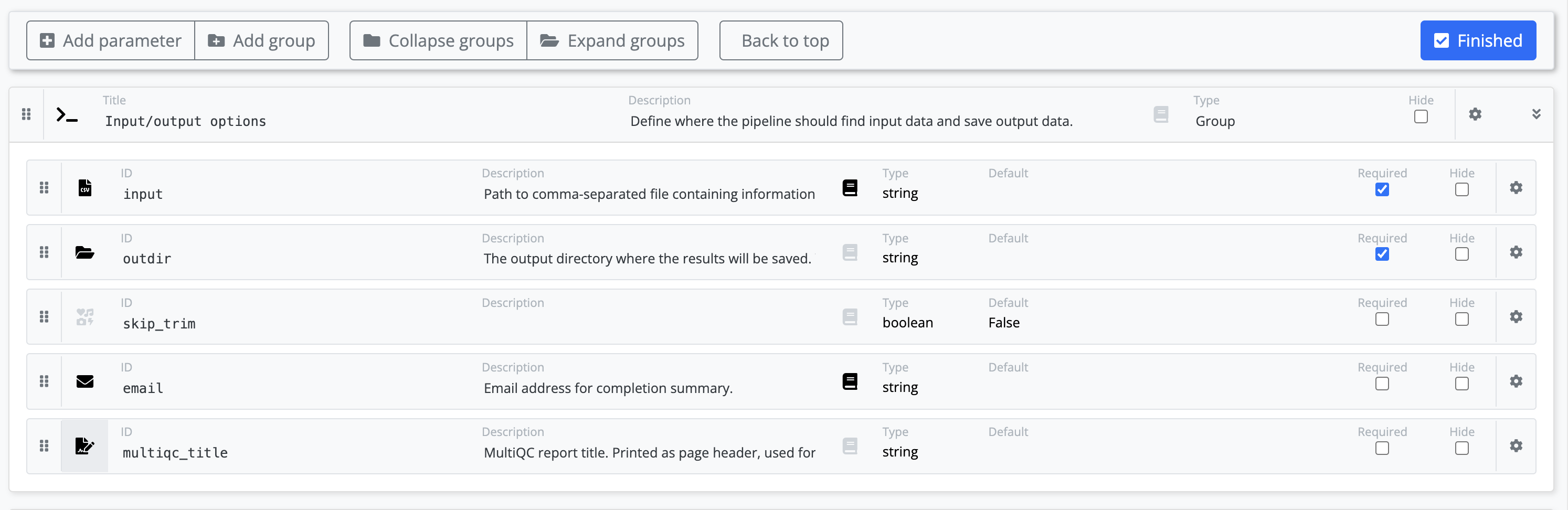
Note
Ungrouped parameters in schema will cause a warning.
Once you have made your edits you can click Finished and all changes will be automatically added to your nextflow_schema.json file.
If you rerun the previous command, the warning should disappear:
nextflow run . -profile test,docker --outdir results --skip_trim
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
executor > local (1)
[6c/c78d0c] process > MYORG_MYFIRSTPIPELINE:MYFIRSTPIPELINE:MULTIQC [100%] 1 of 1 ✔
-[myorg/myfirstpipeline] Pipeline completed successfully-
Takeaway¶
You have added a new parameter to the pipeline, and learned how to use nf-core tools to describe it in the pipeline schema.
What's next?¶
In the next step we will take a look at how we track metadata related to an input file.
Meta maps¶
Datasets often contain additional information relevant to the analysis, such as a sample name, information about sequencing protocols, or other conditions needed in the pipeline to process certain samples together, determine their output name, or adjust parameters.
By convention, nf-core tracks this information as meta maps. These are key-value pairs that are passed into modules together with the files. We already saw this briefly when inspecting the input for seqtk:
If we uncomment our earlier view statement:
| workflows/myfirstpipeline.nf | |
|---|---|
and run the pipeline again, we can see the current content of the meta maps:
You can add any field that you require to the meta map. By default, nf-core modules expect an id field.
Takeaway¶
You know that a meta map is used to pass along additional information for a sample.
What's next?¶
In the next step we will take a look how we can add a new key to the meta map using the samplesheet.
Simple Samplesheet adaptations¶
nf-core pipelines typically use samplesheets as inputs to the pipelines. This allows us to:
- validate each entry and print specific error messages.
- attach information to each input file.
- track which datasets are processed.
Samplesheets are comma-separated text files with a header row specifying the column names, followed by one entry per row. For example, the samplesheet that we have been using during this teaching module looks like this:
sample,fastq_1,fastq_2
SAMPLE1_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R2.fastq.gz
SAMPLE2_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R2.fastq.gz
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,
The structure of the samplesheet is specified in its own schema file in assets/schema_input.json. Each column has its own entry together with information about the column:
"properties": {
"sample": {
"type": "string",
"pattern": "^\\S+$",
"errorMessage": "Sample name must be provided and cannot contain spaces",
"meta": ["id"]
},
"fastq_1": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^\\S+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 1 must be provided, cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
},
"fastq_2": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^\\S+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 2 cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
}
},
"required": ["sample", "fastq_1"]
This validates that the samplesheet has at least two columns: sample and fastq1 ("required": ["sample", "fastq_1"]). It also checks that fastq1 and fastq2 are files, and that the file endings match a particular pattern.
Lastly, sample is information about the files that we want to attach and pass along the pipeline. nf-core uses meta maps for this: objects that have a key and a value. We can indicate this in the schema file directly by using the meta field:
"sample": {
"type": "string",
"pattern": "^\\S+$",
"errorMessage": "Sample name must be provided and cannot contain spaces",
"meta": ["id"]
},
This sets the key name as id and the value that is in the sample column, for example SAMPLE1_PE:
By adding a new entry into the JSON schema, we can attach additional meta information that we want to track. This will automatically validate it for us and add it to the meta map.
Let's add some new meta information, like the sequencer as an optional column:
"properties": {
"sample": {
"type": "string",
"pattern": "^\\S+$",
"errorMessage": "Sample name must be provided and cannot contain spaces",
"meta": ["id"]
},
"sequencer": {
"type": "string",
"pattern": "^\\S+$",
"meta": ["sequencer"]
},
"fastq_1": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^\\S+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 1 must be provided, cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
},
"fastq_2": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^\\S+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 2 cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
}
},
"required": ["sample", "fastq_1"]
We can now run our normal tests with the old samplesheet:
The meta map now has a new key sequencer, that is empty because we did not specify a value yet:
[['id':'SAMPLE1_PE', 'sequencer':[], 'single_end':false], ... ]
[['id':'SAMPLE2_PE', 'sequencer':[], 'single_end':false], ... ]
[['id':'SAMPLE3_SE', 'sequencer':[], 'single_end':true], ... ]
We have also prepared a new samplesheet, that has the sequencer column. You can overwrite the existing input with this command:
This populates the sequencer and we can see it in the pipeline, when viewing the samplesheet channel:
[['id':'SAMPLE1_PE', 'sequencer':'sequencer1', 'single_end':false], ... ]
[['id':'SAMPLE2_PE', 'sequencer':'sequencer2', 'single_end':false], ... ]
[['id':'SAMPLE3_SE', 'sequencer':'sequencer3', 'single_end':true], ... ]
We can comment the ch_samplesheet.view() line or remove it. We are not going to use it anymore in this training section.
Use the new meta key in the pipeline¶
We can access this new meta value in the pipeline and use it to, for example, only enable trimming for samples from a particular sequencer. The branch operator let's us split an input channel into several new output channels based on a selection criteria:
| workflows/myfirstpipeline.nf | |
|---|---|
If we now rerun our default test, no reads are being trimmed (even though we did not specify --skip_trim):
nextflow run . -profile docker,test --outdir results
[- ] process > MYORG_MYFIRSTPIPELINE:MYFIRSTPIPELINE:SEQTK_TRIM -
[5a/f580bc] process > MYORG_MYFIRSTPIPELINE:MYFIRSTPIPELINE:MULTIQC [100%] 1 of 1 ✔
If we use the samplesheet with the sequencer set, only one sample will be trimmed:
nextflow run . -profile docker,test --outdir results --input ../data/sequencer_samplesheet.csv -resume
[47/fdf9de] process > MYORG_MYFIRSTPIPELINE:MYFIRSTPIPELINE:SEQTK_TRIM (SAMPLE2_PE) [100%] 1 of 1 ✔
[2a/a742ae] process > MYORG_MYFIRSTPIPELINE:MYFIRSTPIPELINE:MULTIQC [100%] 1 of 1 ✔
If you want to learn more about how to fine tune and develop the samplesheet schema further, visit nf-schema.
Takeaway¶
You know how to adapt the samplesheet to add new meta information to your files.
What's next?¶
In the next step we will add a module that is not yet in nf-core.
Create a custom module for your pipeline¶
nf-core offers a comprehensive set of modules that have been created and curated by the community. However, as a developer, you may be interested in bespoke pieces of software that are not apart of the nf-core repository or customizing a module that already exists.
In this instance, we will write a local module for the QC Tool FastQE, which computes stats for FASTQ files and print those stats as emoji.
This section should feel familiar to the hello_modules section.
Create the module¶
New module contributions are always welcome and encouraged!
If you have a module that you would like to contribute back to the community, reach out on the nf-core slack or open a pull request to the modules repository.
Start by using the nf-core tooling to create a sceleton local module:
It will ask you to enter the tool name and some configurations for the module. We will use the defaults here:
- Specify the tool name: `Name of tool/subtool: fastqe`
- Add the author name: `GitHub Username: (@<your-name>):`
- Accept the defaults for the remaining prompts by typing `enter`
This will create a new file in modules/local/fastqe.nf that already contains the container and conda definitions, the general structure of the process, and a number of TODO statements to guide you through the adaptation.
Warning
If the module already exists locally, the command will fail to prevent you from accidentally overwriting existing work:
You will notice, that it still calls samtools and the input are bam.
From our sample sheet, we know we have fastq files instead, so let's change the input definition accordingly:
| modules/local/fastqe.nf | |
|---|---|
The output of this tool is a tsv file with the emoji annotation, let's adapt the output as well:
| modules/local/fastqe.nf | |
|---|---|
The script section still calls samtools. Let's change this to the proper call of the tool:
And at last, we need to adapt the version retrieval. This tool does not have a version command, so we will add the release number manually:
| modules/local/fastqe.nf | |
|---|---|
and write it to a file in the script section:
| modules/local/fastqe.nf | |
|---|---|
We will not cover stubs in this training. They are not necessary to run a module, so let's remove them for now:
If you think this looks a bit messy and just want to add a complete final version, here's one we made earlier and we've removed all the commented out instructions:
Include the module into the pipeline¶
The module is now ready in your modules/local folder, but not yet included in your pipeline. Similar to seqtk/trim we need to add it to workflows/myfirstpipeline.nf:
Before:
After:
and call it on our input data:
| workflows/myfirstpipeline.nf | |
|---|---|
Let's run the pipeline again:
In the results folder, you should now see a new subdirectory fastqe/, with the mean read qualities:
Filename Statistic Qualities
sample1_R1.fastq.gz mean 😝 😝 😝 😝 😝 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😉 😉 😜 😜 😜 😉 😉 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😁 😉 😛 😜 😉 😉 😉 😉 😜 😜 😉 😉 😉 😉 😉 😁 😁 😁 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😜 😉 😉 😉 😉 😉 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😛 😜 😜 😛 😛 😛 😚
sample1_R2.fastq.gz mean 😌 😌 😌 😝 😝 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😜 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😉 😜 😉 😉 😜 😜 😉 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😜 😛 😜 😜 😜 😛 😜 😜 😜 😜 😛 😜 😛 😛 😛 😛 😛 😛 😛 😛 😛 😛 😛 😛 😝 😛 😝 😝 😝 😝 😝 😝 😝 😝 😝 😝 😝 😝 😝 😝 😌 😌 😌 😌 😌 😌 😌 😌 😌 😌 😌 😌 😌 😌 😌 😌 😌 😌 😌 😋 😋 😋 😋 😋 😋 😋 😋 😀
Takeaway¶
You know how to add a local module.
And summarise your sequencing data as emojis.
Takeaway¶
You know how to use the nf-core tooling to create a new pipeline, add modulea to it, apply tool and pipeline parameters, and adapt the samplesheet.
What's next?¶
Celebrate and take another break! Next, we'll show you how to take advantage of Seqera Platform to launch and monitor your workflows more conveniently and efficiently on any compute infrastructure.